Announcing wDSSR: The Next-Generation Web Interface to X3DNA-DSSR
Dear 3DNA/DSSR Community,
We are thrilled to announce the official launch of wDSSR (https://web.x3dna-dssr.org/), the powerful new web interface to the X3DNA-DSSR analytical engine.
Developed by Drs. Shuxiang Li and Xiang-Jun Lu and supported by NIH grant R24GM153869, wDSSR represents a major leap forward from our highly popular 2019 Web 3DNA 2.0 framework. While Web 3DNA 2.0 has faithfully served the community for the analysis, visualization, and modeling of 3D nucleic acid structures, wDSSR was built from the ground up to take full advantage of modern web technologies and the latest DSSR backend capabilities.
A Modern, Streamlined Scientific Workflow
We have completely overhauled the user interface to provide a clean, intuitive, and task-driven experience. The core modeling and analysis tools are now seamlessly organized into a logical, single-word scientific workflow: Analyze, Rebuild, Model, Circularize, Mutate, Assemble, and Visualize.
Spotlight Feature: The "Assemble" Module
One of the most exciting upgrades is the newly renamed Assemble tab (formerly "Composite"). This advanced composite model builder allows you to effortlessly construct complex, higher-order models by linking any combination of nucleic acid duplexes or protein-DNA/RNA complexes. You can quickly connect up to six distinct target structures, ranging from simple linked A-DNA and B-DNA duplexes to large, protein-decorated structural assemblies.
Immediate Global Adoption
Although wDSSR has just launched, we are incredibly humbled to share that it is already seeing rapid worldwide adoption! According to recent network infrastructure data, the new interface is actively being used by researchers across North America, South America, Europe, and Asia. Within just a few days, we have recorded active sessions from prestigious institutions around the globe, including:
- The Weizmann Institute of Science in Israel
- Katholieke Universiteit Leuven in Belgium
- Queen's University in Canada
- Universidad Nacional Autonoma de Mexico (UNAM) in Mexico
- Emory University and the Wadsworth Centers Laboratories and Research in the United States
- Jawaharlal Nehru University and the China Education and Research Network in Asia
How to Cite
While a dedicated paper for wDSSR is currently in preparation, researchers should cite the server using its URL (https://web.x3dna-dssr.org/) alongside the 2019 Web 3DNA 2.0 paper and the foundational 2015 DSSR paper. Full details and funding acknowledgements can be found on our newly consolidated About page.
We invite you all to try out the new wDSSR platform! As always, your feedback is invaluable to us, and we encourage you to share your thoughts, questions, and structural models via the newly updated Questions & Feedback link in the wDSSR footer.
Happy modeling!
Dear 3DNA Forum subscribers,
Here are some highlights of recent developments of 3DNA/DSSR:
Note: If you’ve difficulty in accessing the 3DNA homepage, possibly the case from mainland China (as I know it), please visit its duplicate at http://home.x3dna.org. This newsletter is written in Markdown, with a translated HTML version posted on the 3DNA homepage.
3DNA v2.3
The C source code is now available. Since the programs are written in strict ANSI C, 3DNA can be compiled (as is) on any computers/operating systems with a C (or C++) compiler. For user convenience, three binary distributions (with source code under the src/ subdirectory) are provided for Windows, Linux, and Mac OS X. The distributed Windows version works in native Windows (7 and up, via the cmd command-line interface, or ConEMU), MinGW/Msys (Msys2), and Cygwin, in either 32 or 64-bit.
A new set of ‘simple’ base-pair and step parameters was introduced to give ‘intuitive’ numerical values for non-Watson-Crick base pairs and associated steps. See the short communication titled Characterization of base pair geometry in the January 2016 issue of Computational Crystallography Newsletter (CCN).
The fiber program includes a new option, --pauling, for easy generation of Pauling & Corey triplex models of DNA/RNA with arbitrary base sequence. See my blogpost titled Pauling’s triplex model of nucleic acids is available in 3DNA.
Thomas Holder (PyMOL Principal Developer at Schrödinger, Inc.) has built a PyMOL wrapper to 3DNA fiber models. Now generating standard, regular DNA/RNA models in PyMOL is straightforward — thanks, Thomas!
DSSR (Dissecting the Spatial Structure of RNA)
Selected features of DSSR have been incorporated into Jmol (in collaboration with Robert Hanson, Jmol Principal Developer), and PyMOL (in collaboration with Thomas Holder). In Jmol application (via the Console window), one can now, for example, load =1ehz/dssr and then select hairpins; color red to see where the three hairpin loops are in 3D. The Jmol-DSSR web interface makes DSSR-enhanced visualization of nucleic acid structures in Jmol readily accessible to a broad user base, and has been employed in classes for educational purpose. A sample image of DSSR-derived cartoon-block representation via PyMOL is available for PDB entry 5dww, which has a G-quadruplex-duplex interface.
Since the publication of the Nucleic Acids Research paper in 2015, DSSR has been continuously refined and expanded, with a total of 36 new releases (from v1.2.8 to v1.6.4) as of this writing. Notably, the --json option provides DSSR-derived parameters in the simple, structured, and standard JSON format that can be easily parsed. This JSON output format is the (preferred) way for the outside world to interface with DSSR, and the Jmol-DSSR integration is built upon it. The --nmr option allows for batch processing of MODEL/ENDMDL-delineated NMR ensembles or trajectories of molecular dynamics (MD) simulations. Did you know that scripts and data files for reproducing the reported results are available in the DSSR-NAR paper section on the 3DNA Forum?
The User Manual is now 88-page long, covering nevertheless only the most common use cases of what DSSR has to offer. Miss a feature that you would like to have? Maybe it is already there or can be easily implemented in DSSR. Simply ask (on the 3DNA Forum), and I’ll try my best to help.
SNAP (Structures of Nucleic Acid-Protein complexes)
- SNAP aims to consolidate, refine, and significantly extend commonly used functionalities for DNA/RNA-protein structural analysis in one easy-to-use program. Currently in beta testing, SNAP is already fully functional, with features for characterizing the protein-nucleic acid interface and identifying amino acid-base pairing and stacking interactions.
A note for 3DNA/DSSR users in mainland China: It’s a pleasure to see the ~100 registrations on the 3DNA Forum with emails ending in .cn, 163.com, or qq.com etc., mostly from recent years. I’m planning a trip to China in 2017, and I’d be happy to meet some of you for academic exchanges and possible collaborations (学术交流、合作). If you’re interested, let’s get in touch!
Best regards,
Xiang-Jun
—
Dr. Xiang-Jun Lu (律祥俊)
Email: xiangjun@x3dna.org
Web: http://x3dna.org/
Forum: http://forum.x3dna.org/

In 1953, Pauling and Corey published an influential paper, titled A proposed structure for the nucleic acids, in Proc. Natl. Acad. Sci. (PNAS). Key features of the proposed model is summarized in their Letter to Nature, Structure of the Nucleic Acids, published in Nature on February 21, 1953.
We have formulated a structure for the nucleic acids which is compatible with the main features of the X-ray diagram and with the general principles of molecular structure, and which accounts satisfactorily for some of the chemical properties of the substances. The structure involves three intertwined helical polynucleotide chains. Each chain, which is formed by phosphate di-ester groups and linking β-D-ribofuranose or β-D-deoxyribofuranose residues with 3′, 5′ linkages, has approximately twenty-four nucleotide residues in seven turns of the helix. The helixes have the sense of a right-handed screw. The phosphate groups are closely packed about the axis of the molecule, with the pentose residues surrounding them, and the purine and pyrimidine groups projecting radially, their planes being approximately perpendicular to the molecular axis. The operation that converts one residue to the next residue in the polynucleotide chain is rotation by about 105° and translation by 3.4 Å.
This triplex model of nucleic acids, with phosphates in the center and bases on the outside, turned out to be fundamentally flawed. Yet, it played a significant role by prompting Watson and Crick in their discovery of the DNA double helix structure. While I’ve been aware of the Pauling triplex model from long ago, I had not read the original Pauling & Corey PNAS paper. Not surprisingly, I did not know what the triplex structure really looks like, other than some general ideas.
In a recent trip to Rutgers, Dr. Wilma Olson and I discussed the applications of fiber models collected in 3DNA. She drew my attention to the Pauling triplex model, and showed me Table 1 of the PNAS paper (see below), where the atomic coordinates for a nucleic acid repeating unit are listed.

The cylindrical format is the same as that for the fiber models in 3DNA. It thus seems fitting to add this historically significant triplex model to the collection. Googling revealed many interesting historical notes and comments, e.g. The Pauling-Corey Structure of DNA, and a short video Linus Pauling’s triple DNA helix model, 3D animation with basic narration. However, I failed to find a program that I can use to generate such a triplex model with generic base sequence. I decided to add the fiber --pauling option so users can easily create such a triplex model in 3D, just as they do for a classic A- and B-DNA duplex. This process has turned out to be very educational (detailed below), and the end result should be of general interest.
 in Pauling triplex")
- The left 3D image shows the nomenclature of atoms used by Pauling & Corey (see Table 1 above), which is dramatically different from current conventions. As an example, it should be the N1 atom of cytosine (a pyrimidine base), not N3, that is connected to the sugar C1′ atom in nowadays nomenclature. The corrections apply not only to base atoms, but also to the sugar and phosphate groups. The revised atom labeling (as used in the PDB) is illustrated in the 3D image on the right.
- Table 1 corresponds to the ribose sugar since it contains an O2′ atom (see also the figure above). The triplex model constructed would be RNA, but can be ‘converted’ to DNA by simply removing the O2′ atom (see below).
- Only the atomic coordinates for cytosine are listed in Table 1. The 3DNA
mutate_bases program came handy to get the corresponding atomic coordinates for A, G, T, and U. This expansion allows for the generation of Pauling’s triplex models with an arbitrary combination of the five common bases (A, C, G, T, and U).
- With the new
fiber --pauling option, now users can conveniently generate a Pauling’s triplex RNA/DNA model as shown below. Note that the one dash variant -pauling also works fine, with the additional -dna for DNA deoxyribose sugar. The PDB file (Pauling-triplex-mixed.pdb) with mixed DNA sequences can be downloaded, and the corresponding 3D image in top and side views is shown in the following figure.
fiber -pauling triplex-C10C10C10.pdb # default: 10 Cs per strand
fiber -pauling -seq=AAA triplex-A3A3A3.pdb # 3 As per strand
fiber -pauling -seq=AAAA:CCCC:GGGG Pauling-triplex-A4C4G4.pdb
fiber -pauling -seq=ACGGUU,UUGGAC,GGAACC Pauling-triplex-mixed.pdb
fiber --pauling-dna -seq=ACGGTT,TTGGAC,GGAACC Pauling-triplex-DNA.pdb

- With 3DNA’s
find_pair/analyze pair of programs, one can get the structural parameters corresponding to the Pauling triplex model. Not surprising, the repeating dinucleotide along each strand has a twist of 105°, and a rise of 3.4 Å. Notably, the sugar has a C2′-endo conformation.
As of release v2.3-2016sept06, the C source code of the 3DNA software package is available. The code can be found in the $X3DNA/src folder of the distributed tarballs for Linux, Mac OS X, and Windows. Since 3DNA is written in pure ANSI C, it can be compiled without changes on any platform with a modern C compiler.
The original codebase of 3DNA was written around year 2000. Up until v2.3, the infrastructure of 3DNA has remained stable for 16 years. During the time, 3DNA has been widely adopted in other bioinformatics pipelines and cited over 1,500 times. Over the years, I’ve received quite a few requests for 3DNA source code. However, due to complications of various factors (including software licensing), 3DNA had only been distributed in executable forms for the crucial C programs. Now, the C code of 3DNA is finally open source!
As before, users need to register on the 3DNA Forum to download the software. The download page also includes x3dna-v2.0.tar.gz that accompanied the 2008 Nature Protocols paper, and x3dna-v1.5.tar.gz that corresponded to the 2003 Nucleic Acids Research paper. Other than minor revisions to pass strict gcc compiler options, the v1.5 and v2.0 codebases are kept as they were. 3DNA is backward-compatible as far as the key base-pair parameters are concerned. Moreover, between v1.5 and v2.0, the command-line interface stays the same. The two previous versions are released for historical reasons. For example, one may notice some obvious “similarities” between 3DNA v1.5 and RNAView.
The development of DSSR and SNAP will push 3DNA into a brand new version (v3), which contains significant changes in functionality and interface, and is no longer compatible with previous versions. I intend to keep 3DNA v2.3 in a ‘maintenance’ mode: no new features are planed, but bug reports and user questions will be promptly addressed on the 3DNA Forum, as always. Making 3DNA open source should help further prompt its adoptions, and adaptations in structural bioinformatics of nucleic acids.
There are numerous types of software licenses, but none of them seems to be a good fit for my purpose. As a result, I’ve come up with a permissive “citation-ware” license with contents as below:
3DNA is a suite of software programs for the analysis,
rebuilding and visualization of 3-Dimensional Nucleic Acid
structures. Permission to use, copy, modify, and distribute
this suite for any purpose, with or without fee, is hereby
granted, and subject to the following conditions:
At least one of the 3DNA papers must be cited, including the
following two primary ones:
1. Lu, X. J., & Olson, W. K. (2003). "3DNA: a software
package for the analysis, rebuilding and visualization
of three‐dimensional nucleic acid structures." Nucleic
Acids Research, 31(17), 5108-5121.
2. Lu, X. J., & Olson, W. K. (2008). "3DNA: a versatile,
integrated software system for the analysis,
rebuilding and visualization of three-dimensional
nucleic-acid structures." Nature Protocols, 3(7),
1213-1227.
THE 3DNA SOFTWARE IS PROVIDED "AS IS", WITHOUT EXPRESSED OR
IMPLIED WARRANTY OF ANY KIND.
Any 3DNA-related questions, comments, and suggestions are
welcome and should be directed to the open 3DNA Forum
(http://forum.x3dna.org/).
Upon user requests, I’ve recently introduced the --block-color option to DSSR, available as of v1.5.2-2016apr02. As its name implies, the --block-color option facilitate user customization of PyMOL rendered colors of the base rectangular blocks or their edges (e.g., the minor-groove) directly from the command-line. A simple example goes like this: --block-color='A blue; T red', which makes A colored blue and T colored red. As detailed below, the new option is very flexible with regard to the specification of colors, bases, or some edges to highlight. Before that, a little background is in order.
Background info
The DSSR cartoon-block representation follows the color convention of the original 3DNA blocview script, where A is red; C is yellow; G is green; T is blue; and U is cyan. If I remember correctly, the blocview coloring was based on the scheme adopted by the Nucleic Acid Database (NDB). To allow for some flexibility, 3DNA includes a config file named $X3DNA/config/raster3d.par where users can change the RGB values of the corresponding bases. However, I do not know if any user has ever bothered to play around with the configuration file for customized base colors.
Over the years, blocview-generated images have become popular, due to its simplicity, and (maybe more importantly) its endorsement by the NDB and PDB for nucleic acid structures. Via NDB, the blocview-generated images have also been used in RNA FRABASE 2.0 and RNA Structure Atlas. Nevertheless, the blocview script has several dependencies: MolScript for protein or DNA/RNA backbone ribbons, render from Raster3D for rendering, and ImageMagick for image processing. Moreover, the blocview script used by NDB/PDB is (likely to be) based on 3DNA v1.5, the last version before I left Rutgers in 2002.
Over the years, 3DNA has been continuously refined, with significant changes introduced in v2.0 around 2008 to accompany the Nature Protocols paper. Currently at v2.3, the codebase for 3DNA version 2 is in maintenance mode: the software will still be supported with identified bugs fixed, but no more new feature is planned. 3DNA version 3, as represented by DSSR and SNAP, is the way to go.
DSSR has no third-party dependencies
While creating DSSR, I set it as one of the design goals to make the program fully self-contained, without any third-party dependencies. Connections to other tools are clearly delineated via text files. If anything goes wrong, one can easily identify where the problem is. Experience over the past few years has unambiguously proved the effectiveness of this zero-dependency approach. Other than being directly distributed with an operating system, DSSR is the easiest to get up and running. Moreover, DSSR can be easily integrated into other pipelines, including Jmol and PyMOL, among many other bioinformatics tools.
For the cartoon-block representation, DSSR produces .r3d files that can be loaded into PyMOL, mixed and matched with other visualization styles PyMOL has to offer. No more direct dependencies on MolScript, Raster3D, and ImageMagick as is the case for blocview. It is also worth mentioning that DSSR does not need PyMOL to run. DSSR and PyMOL are connected via .r3d files, a process which can be streamlined with the Dssr_block PyMOL plugin.
DSSR releases before v1.5.2-2016apr02 have the color coding of base blocks fixed within the source code, following the default style of blocview. Over the past few months, I’ve received at least two explicit requests on customizing the default colors of DSSR-generated base blocks. The --block-color option has been introduced for this purpose.
Details of the --block-color option
The general format of the option is as follows:
--block-color='id color [; id2 color2 ...]'
id can be A, C, G, T, U, or the degenerated IUPAC code, including R, Y, N etc. See UPAC nucleotide code for details.
id can also be minor, major, upper, bottom, wc-edge to specify one of the six faces of a 3D rectangular block. See Fig.1D of the DSSR paper for details.
id can further be GC, AT, GU, pair, and variants thereof, to specify the colors of the corresponding long base-pair rectangular blocks.
color can be a common name (144 total), as specified in the RGB Color website. For example, red, magenta, light gray etc.
color can also be a single number in the range [0, 1] or [0, 255] to specify a shade of gray. DSSR repeat the number twice to get the RGB triple consisting of the same number.
color can further be a set of three _space_-delimited numbers to specify the RGB triple. Again, the number can be in [0, 1] or [0, 255]. Moreover, the three numbers can be put in square brackets. For example --block-color='A 0 1 1' and --block-color='A [0 1 1]' specify adenine to be colored with RGB triple [0 1 1] (aqua/cyan, corresponding to --block-color='A cyan').
- More than one identity (bases) can be specified, separated by
; (,, :, or | also works). Note: within the PyMOL dssr_block plugin, only | or : can be used as a separator: comma (,) or semicolon (;) cannot be used as a separator within a PyMOL command argument (thanks to Thomas Holder for drawing this point to my attention).
- Case does not matter when specifying
id or color. So either ‘A’ or ‘a’, and ‘blue’ or ‘Blue’ or ‘BLUE’ can be used to make adenine blue: --block-color='a blue'.
Some example usages
While the above description may appears to be quite complicated, the actual usage of the --block-color option is very straightforward. As always, the cases are best made with concrete examples, as shown below using the classic Dickerson B-DNA dodecamer 355d.
# all bases in blue
x3dna-dssr -i=355d.pdb --cartoon-block=orient --block-color='N blue' -o=355d-all-blue.pml
#
# all WC-pairs in red, with the minor-groove edge in 'dim gary'
x3dna-dssr -i=355d.pdb --cartoon-block=orient --block-color='wc-pair red; minor dim gray' -o=355d-pair-minor.pml
#
# thymine (T) in purple, and the upper (+z) face in white
# see Figure below, which shows the two bases in WC-pairs are anti-parallel
x3dna-dssr -i=355d.pdb --cartoon-block=orient --block-color='T purple; upper 1' -o=355d-T-upper.pml
 faces white")
Over the past couple of years, one of the most significant achievements of DSSR has been its integration into Jmol and PyMOL, two widely used molecular graphics programs. None of the projects had been ‘planned’, and I am honored to have the opportunities collaborating directly with Bob Hanson (Jmol) and Thomas Holder (PyMOL). The integrations make salient features of DSSR readily accessible to the Jmol and PyMOL user communities. Moreover, Jmol and PyMOL take different approaches to interoperate with DSSR, and so far they have employed separate features that the program has to offer.
Key features of DSSR
DSSR was implemented in strict ANSI C as a self-contained command-line program. The binaries for common operating systems (Mac OS X, Linux and Windows) are tiny (<1MB), and without runtime dependencies on third-party libraries. DSSR also comes with an extensive PDF user manual.
Since its initial release in early 2013, DSSR has been continuously refined/expanded based on user feedback and my improved knowledge of RNA structures. User questions are always promptly addressed on the public 3DNA Forum. Over the years, DSSR has gradually established itself as an accountable software product.
The small size, zero configuration, extensive features, and robust performance make DSSR ideal to be integrated into other bioinformatics tools.
DSSR and Jmol
From the very beginning, Jmol has been employing a web-service at Columbia University, where all DSSR analyses take place. In addition to the sample DSSR-Jmol web interface, DSSR is also directly accessible from the console (see Fig.1 below). Jmol includes a sophisticated SQL syntax to drill down the various DSSR-derived structure features. Search ‘DSSR’ on the Jmol/JSmol interactive scripting documentation for details.
 Fig. 1 DSSR is available from the Jmol/JSmol console via scripting.
Fig. 1 DSSR is available from the Jmol/JSmol console via scripting.
The initial version of the integration (Jmol v14.2) was facilitated by the DSSR --jmol option to produce a Jmol-specific (e.g., residue id [C]2658:A) plain text output. However, ad hoc text file are rigid and fragile for programs to communicate with. As DSSR had been evolving, changes to existing features or newly added functionality were known to break the established DSSR-Jmol interface. Having to write extra code to maintain the same old --jmol output did not feel right.
JSON (JavaScript Object Notation) came to the rescue! The current DSSR-Jmol integration (Jmol v14.4) takes advantage of JSON, a standard, lightweight data-interchange format. Since JSON is structured, parsing its contents is straightforward. DSSR and Jmol can evolve independently, as always, but they no longer need to worry about touching each other’s toes.
Overall, Jmol has incorporated the most fundamental analysis features of DSSR. The Jmol SQL mini-language is very powerful for selecting arbitrary DSSR parameters. Background information about this collaboration can be found in the blog post Jmol and DSSR.
DSSR and PyMOL
So far, the DSSR-PyMOL integration has focused on visualization, i.e., the cartoon-block schematic representations of DNA/RNA structures. Moreover, instead of relying on a remote DSSR web-service as for Jmol, the PyMOL dssr_block command calls a locally installed DSSR executable for the job. As illustrated in the blogpost DSSR base blocks in PyMOL, interactively, the ‘dssr_block’ command makes it trivial to incorporate the highly effective rectangular blocks into PyMOL.
From early on, 3DNA includes the blocview script (first written in Perl, later converted to Ruby) to generate schematic images in the ‘best view’, by combining block representation of bases with backbone ribbon of proteins or nucleic acids. The script is essentially a glue, calling MolScript, Raster3D, ImageMagick, and several 3DNA utility programs to perform various tasks. With these dependencies, it’s a bit involved to set up blocview. Nevertheless, the resultant images are simple and revealing, and are still being used by NDB and RCSB PDB (among others) as of today.
DSSR does not depend on MolScript and Raster3D, or any other programs to generate .r3d output of rectangular blocks. The schematic blocks can be directly fed into PyMOL, combined with other representations, and ray-traced for high resolution images. The integration of DSSR into PyMOL by the dssr_block command is likely to prompt an even wider adoption of the cartoon-block representation. In this regard, it is well worth noting the news item “dssr_block is a wrapper for DSSR (3dna) and creates block-shaped nucleic acid cartoons” on the main page of PyMOLWiki (see Fig. 2 below). It will certainly bring this neat feature into the attention of many PyMOL users.

Fig. 2 Screenshot of the PyMOLWiki main page (2016-01-27) with ‘dssr_block’ in the news. A sample cartoon-block image of 355d is inserted as an example.
Integration of DSSR analysis results into PyMOL is underway, using the same JSON output. Before long, PyMOL users should be able to have access to the numerous DNA/RNA structural features derived by DSSR as in Jmol, along with the cartoon-block images enabled by dssr_block. Background information about DSSR-PyMOL can be found in blog post Open invitation on writing a DSSR plugin for PyMOL.
Notes
- The DSSR-Jmol and DSSR-PyMOL integrations are two salient examples of what can be achieved via direct collaboration of dedicated scientists with complementary expertise. In addition to benefit the involved projects in particular and the (structural biology) community at large, technical and scientific advances are more likely to be achieved.
- Both projects are still on going, with continued refinements of existing functionality and additions of new features. As an example, it is desirable and likely that Jmol would allow local access to DSSR for efficiency and data privacy.
- JSON is the way to go for connecting DSSR to the outside world. Period. The obsolete
--jmol will be removed from the next release of DSSR (v1.5). The default plain text output is useful for easy comprehension and will stilled be maintained. But do not count on its exact format for computer parsing — occasional changes to existing items are likely, and new features are bound to be added.
- If you’d like to incorporate DSSR into your pipeline and need some customizations of its output, please let me know. It’s always easier to set things right at the source than to fix them downstream. Where practical, I’ll try to implement your requested features, quickly. Working together, we can and will build a better world.
This post is a recap of the recently introduced ‘simple’ base-pair (bp) parameters (Fig. 1) useful for describing non-Waton-Crick pairs, and the highly effective cartoon-block representations of nucleic acid structures. Both features are readily available from 3DNA/DSSR, as detailed here using four examples of representative DNA/RNA structures (Fig. 2). Links to related blog posts are provided at the end.
Note added on Feb. 2, 2016: in fact, this post had been intended to supplement a short communication titled Characterization of base-pair geometry that Dr. Wilma Olson and I recently contributed to the January 2016 issue of Computational Crystallography Newsletter (CCN). That’s why the URL of this post is ‘http://x3dna.org/highlights/CCN-on-base-pair-geometry’ instead of what one would expect from the title. The data files, scripts, images, and linked herein should enable interested users a thorough understanding of the ‘simple’ base-pair parameters. If you have problems in reproducing our reported results, please do not hesitate to let me know (publicly). You are welcome to either leave comments to this post or ask any related questions on the 3DNA Forum.
Six rigid-body parameters

Fig. 1: Schematic diagrams of the six rigid-body parameters commonly used for the characterization of base-pair geometry.
Cartoon-block representations

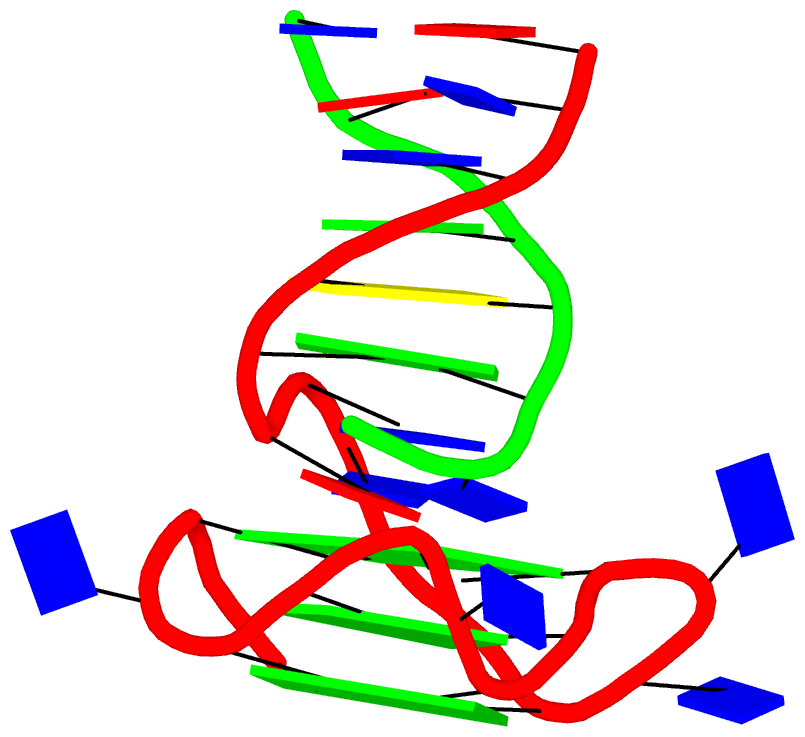
Fig. 2: DSSR-introduced cartoon-block representations of DNA and RNA structures that combine PyMOL cartoon schematics with color-coded rectangular base blocks: A, red; C, yellow; G, green; T, blue; and U, cyan. (A) The Dickerson B-DNA dodecamer solved at 1.4-Å resolution [PDB id: 355d (Shui et al., 1998)], with significant negative Propeller. (B) The Z-DNA dodecamer [PDB id: 4ocb (Luo et al., 2014)], with virtually co-planar C–G pairs at the ends, and noticeable Buckle in the middle. © The GUAA tetraloop mutant of the sarcin/ricin domain from E. coli 23 S rRNA [PDB id: 1msy (Correll et al., 2003)], with large Buckle in the A+C pair, and base-stacking interactions of UAA in the GUAA tetraloop (upper-right corner). (D) The parallel double-stranded poly(A) RNA helix [PDB id: 4jrd (Safaee et al., 2013)], with up to +14° Propeller. The simple, informative cartoon-block representations facilitate understanding of the base interactions in small to mid-sized nucleic acid structures like these. The base identity, pairing geometry, and stacking interactions are obvious.
find_pair 355d.pdb | analyze # 355d.out
x3dna-dssr -i=355d.pdb -more -o=355d-dssr.out
x3dna-dssr -i=355d.pdb --cartoon-block -o=355d.pml
find_pair 4jrd.pdb | analyze # 4jrd.out
x3dna-dssr -i=4jrd.pdb -more -o=4jrd-dssr.out
x3dna-dssr -i=4jrd.pdb --cartoon-block -o=4jrd.pml
find_pair 1msy.pdb | analyze # 1msy.out
x3dna-dssr -i=1msy.pdb -more -o=355d-dssr.out
x3dna-dssr -i=1msy.pdb --cartoon-block -o=1msy.pml
find_pair --symm 4ocb.pdb1 | analyze --symm # 4ocb.out
x3dna-dssr -i=4ocb.pdb1 --symm -more -o=4ocb-dssr.out
x3dna-dssr -i=4ocb.pdb1 --symm --cartoon-block -o=4ocb.pml
Please note the following points:
- The above examples are based on 3DNA
v2.3-2016jan20 and DSSR v1.4.8-2016jan16.
- All data files (including PyMOL ray-traced PNG images used in Fig. 2) are packed into a tarball named Lu-CCN-examples.tar.gz for download.
- For PDB entry 4ocb, the biological unit (with suffix
.pdb1) is used to get a complete duplex structure. The symm option must be specified.
- PDB files are used in the above illustration. In fact, the corresponding mmCIF files (
.cif) also work just fine.
- The DSSR-derived .pml files can be fed into PyMOL for rendering. In addition to the directly generated
*.pml files (e.g., 355d.pml), the PyMOL transformed version (i.e., orient; turn z, -90) are also included, with names *-orient.pml (e.g., 355d-orient.pml). The PNG images (as shown in Fig. 2) are ray-traced using these reoriented pml files for the most extended vertical view.
- The ‘simple’ base-pair parameters for 4jrd is shown below.
This structure contains 10 non-Watson-Crick (with leading *) base pair(s)
----------------------------------------------------------------------------
Simple base-pair parameters based on RC8--YC6 vectors
bp Shear Stretch Stagger Buckle Propeller Opening angle
* 1 A+A -7.96 0.41 -0.03 -13.64 -4.06 -179.47 14.2
* 2 A+A -7.86 0.38 -0.33 -10.20 -3.53 -179.34 10.8
* 3 A+A -7.96 0.43 0.02 -10.15 5.23 179.91 11.4
* 4 A+A -7.95 0.50 0.10 -9.24 8.04 179.15 12.2
* 5 A+A -7.95 0.46 0.08 -7.36 10.12 -179.98 12.5
* 6 A+A -7.97 0.60 0.06 -5.15 12.87 -176.75 13.9
* 7 A+A -7.88 0.66 -0.02 -7.82 11.89 -179.55 14.2
* 8 A+A -7.91 0.56 -0.05 -7.03 13.68 179.22 15.4
* 9 A+A -7.94 0.47 -0.03 -3.78 13.76 -179.24 14.3
* 10 A+A -7.92 0.42 0.10 -3.03 4.34 -178.91 5.3
Related posts
In early 2015, Thomas Holder (the PyMOL Principal Developer at Schrodinger) and I agreed to work together on connecting DSSR to PyMOL. Moreover, we called for the community’s involvement in writing a DSSR plugin for PyMOL and received a few enthusiastic replies. Over the past few months, many significant progresses have been made in DSSR, including an article titled DSSR: an integrated software tool for dissecting the spatial structure of RNA published in Nucleic Acids Research (NAR) and a more streamlined DSSR-Jmol integration based on the --json output.
From the very beginning, Thomas and I had envisioned that the DSSR-PyMOL integration would include two components: one is to bring DSSR-derived RNA/DNA structural features into PyMOL (similar to the DSSR-Jmol interface, funcationality-wise), and the other is to render DSSR’s simple yet informative base-rectangular representations with PyMOL. While the ‘analysis’ component is a work in progress, the ‘visualization’ part is ready for the community to take advantage of.
Thomas has written a Python script named dssr_block.py. When the script is run in PyMOL, it adds the “dssr_block” command. The dssr_block.py script is less than 100 lines including documentation, with the real code taking no more than half of the total line number. The detailed documentation section (with two examples), when condensed, is as follows:
DESCRIPTION
Create a nucleid acid cartoon with DSSR
USAGE
dssr_block [selection [, state [, block_file [, block_depth [, name [, exe]]]]]]
ARGUMENTS
selection = str: atom selection {default: all}
state = int: object state (0 for all states) {default: -1, current state}
block_file = face|edge|wc|equal|minor|gray {default: face}
block_depth = float: thickness of rectangular blocks {default: 0.5}
name = str: name of new CGO object {default: dssr_block##}
exe = str: path to "x3dna-dssr" executable {default: x3dna-dssr}
EXAMPLE
fetch 1ehz, async=0
as cartoon
dssr_block
set cartoon_ladder_radius, 0.1
set cartoon_ladder_color, gray
set cartoon_nucleic_acid_mode, 1
# multi-state
fetch 2n2d, async=0
dssr_block 2n2d, 0
set all_states
Download the dssr_block.py script into a folder (directory) of your choice. Within PyMOL command window, type:
run dssr_block.py # to make the 'dssr_block' command avaible
help dssr_block # to get the help message, with contents shown above
The resultant cartoon-block image for running the documented commands (except for the additional orient command for best view) for case 1ehz is shown in Fig. 1 below.
")
Fig. 1: Cartoon-block image generated by dssr_block.py for PDB entry 1ehz (yeast phenylalanine tRNA)
For the NMR ensemble 2n2d, the corresponding image (after running orient) is illustrated in Fig. 2 as follows:
")
Fig. 2: Cartoon-block image generated by dssr_block.py for PDB entry 2n2d (an NMR ensemble).
In addition to the default settings, DSSR offers quite a few variations for the size and coloring of rectangular blocks, as demonstrated in Fig.3. The main settings are through the block_file option in PyMOL (note the underscore), corresponding to DSSR --block-file (or --block_file). The corresponding PyMOL commands are also listed for your reference. You can easily play around with the various styles interactively in PyMOL by toggling objects (dssr_block##) on or off. Enjoy!
")
Fig. 3: Cartoon-block image generated by dssr_block.py for PDB entry 355d (the Dickerson B-DNA dodecamer).
Fig. 3 is created with the following PyMOL commands:
reinitialize
fetch 355d, async=0
bg_color white
as cartoon
orient
turn z, -90
turn y, 180
set cartoon_ladder_mode, 1
set cartoon_ladder_radius, 0.1
set cartoon_ladder_color, black
set cartoon_tube_radius, 0.5
set cartoon_nucleic_acid_mode, 1
set cartoon_color, gold
dssr_block 355d # default base blocks in solid color
dssr_block block_file=edge # rectangular blocks in wireframe (black)
dssr_block block_file=face+edge # solid color with outline
dssr_block block_file=equal # bases blocks in equal size
dssr_block block_file=minor # with minor-groove colord black
dssr_block block_file=wc # Watson-Crick base pairs in long bp blocks
dssr_block block_file=wc-minor # Watson-Crick pairs + minor-groove edge
dssr_block block_file=gray # rectangular blocks all in gray
dssr_block block_depth=1.8 # with increased thickness
Notes
- The
dssr_block.py script described here is the original version Thomas communicated to me. Current version of this script and related topics can be found in the Dssr block PyMOLWiki page.
- For this script to work, DSSR needs to be installed and
x3dna-dssr in the PATH.
- In PyMOL,
set cartoon_nucleic_acid_mode, 1 employs C3′ instead of the default P (‘mode 0’) for the smooth backbone trace. Since 5′ terminal phosphate groups are normally not available from X-ray crystal structures (e.g., 355d), ‘mode 1’ is used to avoid orphan base blocks from the backbone trace.
With the foundation laid by the previous two posts on Fitting of base reference frame and Automatic identification of nucleotides, we can now get into the details on how the ‘simple’ base-pair (bp) parameters are derived. To make the point clear, I am using two concrete examples from the yeast phenylalanine tRNA (PDB id: 1ehz): the first pair is 2MG10+G45, of type M+N (shortened to g+G) in 3DNA/DSSR; and the second example is a Watson-Crick pair U6–A67, of type M–N (shortened to U–A).
Pair 2MG10+G45 (g+G, of type M+N, see Fig. 1)
Base reference frames
")
Fig. 1: Base pair 2MG10+G45 (g+G) of type M+N in yeast phenylalanine tRNA 1ehz
In the original coordinate system (as in 1ehz.pdb downloaded from the RCSB PDB), the base-reference frames for 2MG10 and G45 are:
# base reference frame of 2MG10
{
"rsmd": 0.018218,
"origin": [65.696016, 45.134944, 18.125044], # o1
"x_axis": [0.690346, 0.713907, -0.117302], # x1
"y_axis": [-0.706849, 0.700116, 0.101003], # y1
"z_axis": [0.154232, 0.013188, 0.987947] # z1
}
# base reference frame of G45
{
"rsmd": 0.025865,
"origin": [70.584399, 50.526567, 17.229626], # o2
"x_axis": [0.818521, 0.49914, -0.284399], # x2
"y_axis": [-0.574112, 0.728382, -0.373973], # y2
"z_axis": [0.020486, 0.469381, 0.882758] # z2
}
The base-pair reference frame
Since dot(z1, z2) = 0.88 (positive), this pair is of type M+N in 3DNA/DSSR. The ‘mean’ z-axis of the pair is the average of z1 and z2, which is z = [0.090069, 0.248769, 0.964366] (normalized). This is the z-axis of the bp frame, as in 3DNA/DSSR.
The ‘long’ axis employs RC8 (purines) and YC6 (pyrimidines) base atoms. Here 2MG10 and G45 are all purines, so the following two C8 atoms are used:
# C8 atoms of 2MG10 and G45 in 1ehz
HETATM 208 C8 2MG A 10 62.199 48.621 18.635 1.00 40.38 C
ATOM 987 C8 G A 45 67.772 54.149 15.386 1.00 40.45 C
The vector from C8 of G45 to C8 of 2MG10 is:
y0 = [62.199 48.621 18.635] - [67.772 54.149 15.386]
= [-5.573 -5.528 3.249]
Normally, y0 and z-axis are not orthogonal. Here they have an angle of ~81º. The orthogonal component of y0 with reference to the z-axis, when normalized, is the y-axis:
y = [-0.676751, -0.695120, 0.242520]
The x-axis is defined by the right-handed rule:
x = [-0.730682, 0.674479, -0.105746]
Overall, the orthonormal x-, y- and z-axes of the pair defined thus far are:
x = [-0.730682, 0.674479, -0.105746]
y = [-0.676751, -0.695120, 0.242520]
z = [0.090069, 0.248769, 0.964366]
Derivation of the six ‘simple’ base-pair parameters (Fig. 2)
Fig. 2: Schematic diagram of six rigid-body base-pair parameters
Propeller is the ‘torsion’ angle of z2 to z1 with reference to the y-axis, and is calculated using the method detailed in the blog post How to calculate torsion angle?. Here Propeller is: -24.24º. Similarly, Buckle is defined as the ‘torsion’ angle of z2 to z1 with reference to the x-axis, and is -14.81º. Opening is defined as the angle from y2 to y1 with reference to the z-axis, and is: 13.32º.
The corresponding translational parameters are simply projects of the o2 to o1 vector onto the x-, y- and z-axis, respectively. Here, they have values:
d = o1 - o2 = [-4.888383, -5.391623, 0.895418]
Shear = dot(d, x) = -0.16
Stretch = dot(d, y) = 7.27
Stagger = dot(d, z) = -0.92
‘Corrections’ of Buckle and Propeller
Base-pair non-planarity is due to the following three parameters: Buckle, Propeller, and Stagger. In particular, Buckle and Propeller cause the two bases to be non-parallel, the most noticeable characteristic of a pair. These two angular parameters are well-documented in literature, even among the canonical Watson-Crick base pairs. In 3DNA/DSSR, the angle between the two base normal vectors (in range [0, 90º]) is related to Buckle and Propeller with the formula:
interBase-angle = sqrt(Buckle^2+Propeller^2)
For the 2MG10+G45 pair, the angle between z1 and z2 is 28.18º, and sqrt(Buckle^2+Propeller^2) = 28.405º. So the following ‘corrections’ are made:
Buckle = -14.81 * 28.18 / 28.405 = -14.69
Propeller = -24.24 * 28.18 / 28.405 = -24.05
Overall, the ‘corrections’ have only small influence on the numerical values of the reported Buckle and Propeller parameters. It is ‘sensible’ that the ‘simple’ parameters have the property interBase-angle = sqrt(Buckle^2+Propeller^2), just as the original 3DNA/DSSR bp parameters.
Now, the six ‘simple’ bp parameters for 2MG10+G45, reported in 3DNA analyze program as of v2.3-2016jan01 are:
Simple base-pair parameters based on YC6-RC8 vectors
bp Shear Stretch Stagger Buckle Propeller Opening angle
* 1 g+G -0.16 7.27 -0.92 -14.69 -24.05 13.32 28.2
The corresponding local bp parameters as originally reported by 3DNA/DSSR are as follows. Note the significant differences in Shear vs. Stretch, and Buckle vs. Propeller in the two sets of bp parameters. On the other hand, Stagger is identical and Opening should be quite close, by definition. Due to the similarity in Stagger and Opening, DSSR only reports four ‘simple’ parameters (i.e., Shear, Stretch, Buckle, and Propeller).
Local base-pair parameters
bp Shear Stretch Stagger Buckle Propeller Opening
1 g+G -7.21 -0.97 -0.92 25.58 -11.83 13.07
Base-pair U6–A67 (Watson-Crick U–A, of type M–N, see Fig. 3)
")
Fig. 3: Base pair U6–A67 (U–A) of type M–N in yeast phenylalanine tRNA 1ehz
Base reference frames
In the original coordinate system (as in 1ehz.pdb downloaded from the RCSB PDB), the base-reference frames for U6 and A67 are:
# base reference frame of U6 (white in Fig. 3)
{
"rsmd": 0.010835,
"origin": [60.441988, 48.83479, 41.242523], # o1
"x_axis": [0.28491, 0.503019, 0.815965], # x1
"y_axis": [0.887155, -0.460753, -0.025726], # y1
"z_axis": [0.363018, 0.731217, -0.577529] # z1
}
# base reference frame of A67 (colored yellow in Fig. 3)
{
"rsmd": 0.01992,
"origin": [60.578326, 48.823104, 41.154211], # o2
"x_axis": [0.034097, 0.205538, 0.978055], # x2
"y_axis": [-0.90687, 0.417653, -0.056155], # y2
"z_axis": [-0.420029, -0.885054, 0.200637] # z2
}
The base-pair reference frame
Since dot(z1, z2) = -0.92 (negative), this pair is of type M–N in 3DNA/DSSR. The y- and z-axis are thus reversed (corresponding to a 180º rotation around the x-axis) to align z2 with z1.
# base reference frame of A67, with y- and z-axes reversed
{
"origin": [60.578326, 48.823104, 41.154211], # o2
"x_axis": [0.034097, 0.205538, 0.978055], # x2
"y_axis": [0.90687, -0.417653, 0.056155], # y2 -- reversed
"z_axis": [0.420029, 0.885054, -0.200637] # z2 -- reversed
}
Thereafter, the procedure is similar to the one for the M+N type above. Note here U6 is a pyrimidine, so its C6 atom is used. The final results are:
# C6 atom of U6 and C8 atom A67 in 1ehz
ATOM 132 C6 U A 6 64.926 46.497 41.084 1.00 35.72 C
ATOM 1457 C8 A A 67 56.129 50.866 40.893 1.00 40.04 C
#---------
y0 = [64.926 46.497 41.084] - [56.129 50.866 40.893]
= [8.797 -4.369 0.191]
x = [0.160777, 0.363836, 0.917482]
y = [0.902274, -0.430972, 0.012793]
z = [0.400064, 0.825764, -0.397570]
The six ‘simple’ and original base-pair parameters
Simple base-pair parameters based on YC6-RC8 vectors
bp Shear Stretch Stagger Buckle Propeller Opening angle
1 U-A 0.06 -0.13 -0.08 -0.59 -23.71 5.39 23.7
# ------------
Local base-pair parameters
bp Shear Stretch Stagger Buckle Propeller Opening
1 U-A 0.06 -0.13 -0.08 -0.63 -23.71 5.50
As can be seen, for Watson-Crick pairs, the ‘simple’ and the original bp parameters are very similar.
Special notes on the ‘simple’ base-pair parameters

- For the most common Watson-Crick pairs, the newly introduced ‘simple’ bp parameters match those of the original 3DNA/DSSR parameters very well (as shown by the U6–A67 pair). For non-canonical pairs, significant differences in Shear, Stretch, Buckle and Propeller are expected (as illustrated by the 2MG10+G45 pair). The differences come from the divergent definitions of the bp reference frame, which is distinct for each type of non-canonical pairs.
- Only the original 3DNA/DSSR six bp parameters can be used for exact reconstruction (with the 3DNA
rebuild program) of the corresponding bp geometry. The ‘simple’ bp parameters are for description only, and they could be more intuitive than the original 3DNA/DSSR counterparts. They complement, buy by no means replace, the classic “local” bp parameters. The term ‘simple’ is used to distinguish the new from the original closely related, yet quite different bp parameters.
- As details for the 2MG10+G45 pair, several ad hoc decisions are made in deriving the ‘simple’ bp parameters. For example, instead of using RC8–YC6 to define the y-axis, one can also use RN9–YN1 (as did by Richardson). Each such choice will lead (slightly) different numerical values, depending on the type of the non-canonical pairs. In some cases, Buckle and Propeller could differ by several degrees. Since RC8 and YC6 atoms lie near the ‘center’ of purines and pyrimidines, they are used to define the y-axis (by default). DSSR has provisions of selecting RN9–YN1, as well as a couple of other choices, for the definition of the y-axis.
- When the M+N pair is counted as N+M, Shear, Stretch, Buckle, and Propeller remain the same, but Stagger and Opening reverse their signs. For example, here are the results of 2MG10+G45 vs. G45+2MG10:
# 2MG10+G45
Simple base-pair parameters based on YC6-RC8 vectors
bp Shear Stretch Stagger Buckle Propeller Opening angle
* 1 G+g -0.16 7.27 0.92 -14.69 -24.05 -13.32 28.2
# Reverse the order: treated as G45+2MG10
Simple base-pair parameters based on YC6-RC8 vectors
bp Shear Stretch Stagger Buckle Propeller Opening angle
* 1 g+G -0.16 7.27 -0.92 -14.69 -24.05 13.32 28.2
- When the M–N pair is counted as N–M, Stretch, Stagger, Propeller, and Opening remain the same, but Shear and Buckle reverse their signs. For example, here are the results of U6–A67 vs. A67–U6:
# U6–A67
Simple base-pair parameters based on YC6-RC8 vectors
bp Shear Stretch Stagger Buckle Propeller Opening angle
1 U-A 0.06 -0.13 -0.08 -0.59 -23.71 5.39 23.7
# Reverse the order: treated as A67–U6
Simple base-pair parameters based on YC6-RC8 vectors
bp Shear Stretch Stagger Buckle Propeller Opening angle
1 A-U -0.06 -0.13 -0.08 0.59 -23.71 5.39 23.7
Related posts
Once a nucleotide (nt) is identified, and matched to A (C, G, T, U) for the standard case or a (c, g, t, u) for a modified one, 3DNA/DSSR performs a least-squares fitting procedure to locate the base reference frame in three-dimensional space. The basic idea is very simple and widely applicable. The algorithm constitutes one of the key components of 3DNA/DSSR. As always, the details can be most effectively illustrated with a worked example. Using G1 in the yeast phenylalanine tRNA (PDB id: 1ehz) as an example, the atomic coordinates of its nine base-ring atoms are:
# G1, nine base-ring atoms for ls-fitting
ATOM 14 N9 G A 1 51.628 45.992 53.798 1.00 93.67 N
ATOM 15 C8 G A 1 51.064 46.007 52.547 1.00 92.60 C
ATOM 16 N7 G A 1 51.379 44.966 51.831 1.00 91.19 N
ATOM 17 C5 G A 1 52.197 44.218 52.658 1.00 91.47 C
ATOM 18 C6 G A 1 52.848 42.992 52.425 1.00 90.68 C
ATOM 20 N1 G A 1 53.588 42.588 53.534 1.00 90.71 N
ATOM 21 C2 G A 1 53.685 43.282 54.716 1.00 91.21 C
ATOM 23 N3 G A 1 53.077 44.429 54.946 1.00 91.92 N
ATOM 24 C4 G A 1 52.356 44.836 53.879 1.00 92.62 C
The corresponding nine base-ring atoms of G in its standard base reference frame are listed below. See Table 1 of the report A Standard Reference Frame for the Description of Nucleic Acid Base-pair Geometry, and file Atomic_G.pdb distributed with 3DNA ($X3DNA/config/Atomic_G.pdb). In DSSR, the content has been integrated into the source code to make the program self-contained.
# G in standard base reference frame
ATOM 2 N9 G A 1 -1.289 4.551 0.000
ATOM 3 C8 G A 1 0.023 4.962 0.000
ATOM 4 N7 G A 1 0.870 3.969 0.000
ATOM 5 C5 G A 1 0.071 2.833 0.000
ATOM 6 C6 G A 1 0.424 1.460 0.000
ATOM 8 N1 G A 1 -0.700 0.641 0.000
ATOM 9 C2 G A 1 -1.999 1.087 0.000
ATOM 11 N3 G A 1 -2.342 2.364 0.001
ATOM 12 C4 G A 1 -1.265 3.177 0.000
A least-squares fitting of the standard onto the experimental set of base-ring atoms defines the base reference frame (Fig. 1). The information is available via the following commands:
# find_pair -s 1ehz.pdb # in file 'ref_frames.dat'
... 1 G # A:...1_:[..G]G
53.7571 41.8678 52.9303 # origin
-0.2589 -0.2496 -0.9331 # x-axis
-0.5430 0.8365 -0.0731 # y-axis
0.7988 0.4878 -0.3521 # z-axis
# --------
# x3dna-dssr -i=1ehz.pdb --json | jq .nts[0].frame
{
rsmd: 0.008,
origin: [53.757, 41.868, 52.93],
x_axis: [-0.259, -0.25, -0.933],
y_axis: [-0.543, 0.837, -0.073],
z_axis: [0.799, 0.488, -0.352]
}

Fig. 1: G1 in tRNA 1ehz, with base reference frame attached
Please note the following subtle points:
- The standard base (
Atomic_G.pdb) is already set in its reference frame: the _z_-coordinates are virtually zeros, _y_-coordinates are positive, the atoms along the minor-groove edge have negative _x_-coordinates, as can be visualized clearly from the attached coordinate frame. In 3DNA, the five standard standard bases are in stored in files Atomic_[ACGTU].pdb, and the corresponding modified ones are in Atomic_[acgtu].pdb. For simplicity, Atomic_A.pdb and Atomic_a.pdb are the same by default, as are the other four cases.
- The translation and rotation of the least-squares fitting process define the experimental base reference frame (for G1 in the above example), and its three axes are orthonormal by definition.
- By design, the base rings of
Atomic_A.pdb and Atomic_G.pdb match each other closely (see below), as are the pyrimidines bases. The least-square fitted root-mean-square deviation (rmsd) of the nine base-ring atoms between standard A and G is only 0.04 Å. Fitting the standard A (instead of G) onto G1 of 1ehz leads to a base reference frame that is essentially indistinguishable from the one above (see below). This feature shows that any ambiguity in assigning modified purines to A or G, or pyrimidines to C, T, or U causes no notable differences in 3DNA/DSSR results.
Comparison of base-ring atomic coordinates in standard G and A
Atomic_G.pdb Atomic_A.pdb
N9 G -1.289 4.551 0.000 | N9 A -1.291 4.498 0.000
C8 G 0.023 4.962 0.000 | C8 A 0.024 4.897 0.000
N7 G 0.870 3.969 0.000 | N7 A 0.877 3.902 0.000
C5 G 0.071 2.833 0.000 | C5 A 0.071 2.771 0.000
C6 G 0.424 1.460 0.000 | C6 A 0.369 1.398 0.000
N1 G -0.700 0.641 0.000 | N1 A -0.668 0.532 0.000
C2 G -1.999 1.087 0.000 | C2 A -1.912 1.023 0.000
N3 G -2.342 2.364 0.001 | N3 A -2.320 2.290 0.000
C4 G -1.265 3.177 0.000 | C4 A -1.267 3.124 0.000
Comparison of G1 (1ehz) base reference frame derived using standard G or A
Atomic_G.pdb | Atomic_A.pdb
53.7571 41.8678 52.9303 # origin | 53.7286 41.9276 52.9482 # origin
-0.2589 -0.2496 -0.9331 # x-axis | -0.2562 -0.2540 -0.9327 # x-axis
-0.5430 0.8365 -0.0731 # y-axis | -0.5444 0.8352 -0.0780 # y-axis
0.7988 0.4878 -0.3521 # z-axis | 0.7988 0.4878 -0.3522 # z-axis
Related topics:
{kind=link}
{kind=link}