A recent thread on the 3DNA Forum discussed 'Rebuilding Z-DNA' by extending an existing structure. The 3DNA rebuild program allows users to generate DNA or RNA structures with any user-specific sequence and corresponding base-pair/step parameters. This process is rigorous for atomic coordinates of base (and C1') atoms: running analyze on the rebuilt structure will yield the same set of parameters that users initially input. For more details, see the 2003 3DNA paper, the 2015 DSSR paper, and the DSSR User Manual.
The challenge lies in modeling the backbones. For right-handed A- or B-form DNA, users can build full-atomic models with canonical backbone conformations of C3'-endo or C2’-endo sugar conformations and anti glycosidic bonds. However, left-handed Z-DNA has unique structural features—such as syn-G, CpG, and GpC dinucleotides as building blocks instead of single nucleotides—that are not fully addressed by the 3DNA rebuild program.
DSSR (Pro version or the Academic v2.5.2) offers a solution by providing tools to build extended Z-DNA structures with proper backbones. The commands are as follows:
x3dna-dssr -i=1qbj.pdb1 --select-chains='D E' --delete-water -o=model.pdb
x3dna-dssr tasks -i=model.pdb --frame-pair=last -o=model1-ref-last.pdb
# poly d(GC) : poly d(GC)
x3dna-dssr fiber --z-dna --repeat=1 -o=conn.pdb
x3dna-dssr tasks -i=conn.pdb --frame-pair=first --remove-pair -o=ref-conn.pdb
x3dna-dssr tasks --merge-file='model1-ref-last.pdb ref-conn.pdb' -o=temp1.pdb
x3dna-dssr tasks -i=temp1.pdb --frame-pair=last --remove-pair -o=temp2.pdb
x3dna-dssr tasks -i=model.pdb --frame-pair=first -o=model1-ref-first.pdb
x3dna-dssr tasks --merge-file='temp2.pdb model1-ref-first.pdb' -o=duplicate-model.pdb
x3dna-dssr --order-residue -i=duplicate-model.pdb -o=temp3.pdb --po-bond=3.6
x3dna-dssr --renumber-residue -i=temp3.pdb -o=temp4.pdb
x3dna-dssr --connect-file -i=temp4.pdb -o=1qbj-duplicate.pdb --po-bond=3.6The logic behind these commands is very straightforward, but technical details may look a bit complex for the uninitiated:
- The first command extracts the Z-DNA duplex consisting of chains D and E from PDB entry
1qbj.pdb1(the first biological unit) and remove water molecules (model.pdb). The Z-DNA duplex has sequence: CGCGCG/CGCGCG. - The next command sets the Z-DNA duplex (
model.pdb) into the reference frame of the last base pair, i.e., G-C (model1-ref-last.pdb). - The
fibermodel consists of the GpC dinucleotide step (conn.pdb), which is then set into the reference frame of the first base pair (G-C). The first G-C pair is removed from the coordinate fileref-conn.pdbwhich consists of only one C-G pair. - The two PDB files,
model1-ref-last.pdbandref-conn.pdb, share a common reference frame and are merged into a single PDB file (temp1.pdb). - The merged PDB file (
temp1.pdb) is then set into the reference frame of last base pair(i.e., C-G) which is removed from the resulting coordinate file (temp2.pdb). Now the job of the GpC fiber connector is done. - The Z-DNA duplex (
model.pdb) is once again set into the reference frame of the first base pair (i.e., C-G), leading to the coordinate filemodel1-ref-first.pdb. - The two PDB files,
temp2.pdbandmodel1-ref-first.pdb, both consist of the same Z-DNA duplex but are in different orientations. They now share a common reference frame and are merged into the extended Z-DNA duplex (1qbj-duplicate.pdb). - The last three commands (with options
--order-residue,--renumber-residue,--connect-file) are bookkeeping steps to ensure proper order and numbering of nucleotides along each chain, and generate theCONECTrecord for smooth view in PyMOL.
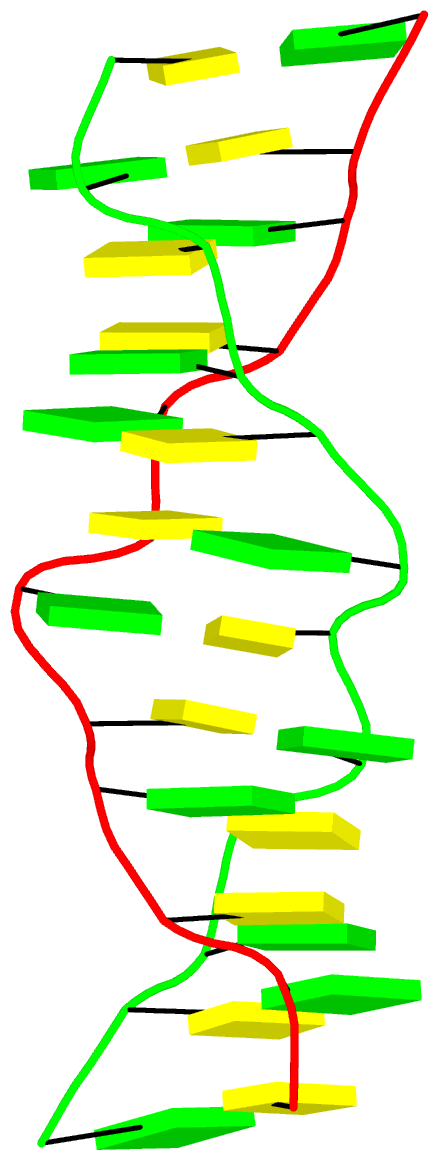
The final PDB coordinate file (1qbj-duplicate.pdb) can be downloaded, and visualized in DSSR-enabled cartoon-block representation as below: