Background and motivation
In late 2021, I came across the thread titled "create a 26 bp RNA from a 13 bp system" on the PyMOL mailing list. The thread began with a user asking:
I have an RNA duplex with 13 base-pairs (attached). Is it possible to duplicate this system and then fuse the two molecules to create a 26 base-pair long system using the pymol.
The message is both concise and clear. The attached 13 base-pair RNA duplex (named model.pdb) makes the task easier to understand. An expert PyMOL user responded quickly, providing a set of suggested PyMOL commands along with warnings about the complexity of the task.
No, not automatically. Your RNA is very distorted from the standard A-form. I doubt any modeling program can accurately extend such a distorted helix. Maybe someone else will prove me wrong. ... You can align the terminal base pairs manually through a series of commands. If you try by dragging one copy relative to another, you will wind up pulling out all of your hair. The commands and patience will keep you out of the mad house.
DSSR offers unique capabilities to automatically manipulate nucleic acid structures. It also enables the duplication of an RNA duplex, as specifically requested by the original poster. In my initial response to the thread, I provided a DSSR-based solution for duplicating the RNA duplex without detailed explanations, aiming to confirm whether the result met the user's needs. The feedback was positive, as indicated below:
Thanks for proving me wrong. Congratulations on your duplicated model! Please share the commands that you used with DSSR to generate the duplicated helix. --- from the PyMOL responder
Thanks a lot for your help. The model you have duplicated is exactly what I am looking for (checked it with VMD). Unfortunately I do not have access to DSSR-Pro. Is there any way that I can reproduce your procedure with x3dna-dssr? I need to create different numbers of duplicates (2,4,6,5,8) for different systems and this will be very helpful. --- from the original poster
During that period (near the end of 2021), I was facing a funding gap. To address this challenge, we decided to license DSSR through Columbia Technology Ventures (CTV) and introduced a Pro version of DSSR for commercial users and academic institutions, providing advanced modeling features and dedicated support. Note that DSSR Pro Academic licenses entail a one-time fee of $1,020. The software can be installed on Windows, macOS, or Linux. While not explicitly included in the license agreement, I provide direct support to Pro license users via email, phone, or Zoom whatever convenient to help address their issues. I care about user experience, especially for those who invest in the Pro version.
Following user feedback, I shared detailed instructions on duplicating an RNA duplex using DSSR Pro. Gratefully, the original poster purchased a DSSR Pro Academic license and successfully duplicated the RNA helix. Later, we communicated via email to assist with other related tasks. This experience underscored the importance of engaging with the scientific community and addressing user needs to drive software development and adoption.
Detailed instructions
With funding from grant R24GM153869, I have transferred many DSSR Pro features into the free DSSR Academic version to better serve the scientific community. Included below are detailed step-by-step commands script for duplicating an RNA duplex using either DSSR Pro or the free DSSR Academic v2.5.2. The script runs instantaneously in a terminal window.
x3dna-dssr tasks -i=model.pdb --frame-pair=last -o=model1-ref-last.pdb
x3dna-dssr fiber --seq=GG --rna-duplex -o=conn.pdb
x3dna-dssr tasks -i=conn.pdb --frame-pair=first --remove-pair -o=ref-conn.pdb
x3dna-dssr tasks --merge-file='model1-ref-last.pdb ref-conn.pdb' -o=temp1.pdb
x3dna-dssr tasks -i=temp1.pdb --frame-pair=last --remove-pair -o=temp2.pdb
x3dna-dssr tasks -i=model.pdb --frame-pair=first -o=model1-ref-first.pdb
x3dna-dssr tasks --merge-file='temp2.pdb model1-ref-first.pdb' -o=duplicate-model.pdb
x3dna-dssr --order-residue -i=duplicate-model.pdb -o=temp3.pdb
x3dna-dssr --renumber-residue -i=temp3.pdb -o=temp4.pdb
x3dna-dssr --connect-file -i=temp4.pdb -o=RNA-duplicate.pdbThe procedure is essentially the same as the one used in "Building extended Z-DNA structures with backbones using DSSR". For completeness, I have included detailed explanations for each step here as well.
-
Setting Up the Reference Frame:
- The first command places the 13 base-pair RNA duplex (
model.pdb) into the reference frame of its last base pair, resulting inmodel1-ref-last.pdb.
- The first command places the 13 base-pair RNA duplex (
-
Creating the Fiber Connector:
- The
fibermodel is constructed using an RNA duplex (--rna-duplex) with the sequence GG on the leading strand (conn.pdb). This connector is oriented into the reference frame of its first base pair. - The first base pair is removed. Thus, the resulting coordinate file,
ref-conn.pdb, contains only one pair. - Note: The sequence GG serves as a placeholder. It can be replaced with any other two bases: for instance, changing
--seq=GGto--seq=AA. Moreover, using--seq=GA10Gallows for creating a linker with 10 adenines.
- The
-
Merging PDB Files:
- The two PDB files,
model1-ref-last.pdbandref-conn.pdb, share a common reference frame and are merged into a single file namedtemp1.pdb.
- The two PDB files,
-
Adjusting the Reference Frame:
- The merged file (
temp1.pdb) is then aligned with the last base pair, which is subsequently removed to producetemp2.pdb. This completes the role of the GG fiber connector.
- The merged file (
-
Reorienting the RNA Duplex:
- The original 13 base-pair RNA duplex (
model.pdb) is reoriented into the reference frame of its first base pair, generatingmodel1-ref-first.pdb.
- The original 13 base-pair RNA duplex (
-
Final Merging:
- The two PDB files,
temp2.pdbandmodel1-ref-first.pdb, contain identical 13 base-pair RNA duplexes but in different orientations. They are merged into a single file (duplicate-model), establishing the final duplicated RNA structure.
- The two PDB files,
- Bookkeeping for Visualization:
- The last three commands utilize options (
--order-residue,--renumber-residue,--connect-file) to prepare the structure for visualizing backbone connectivity in PyMOL. Specifically, it solves the reported issue that "PyMOL does not generate the cartoon representation for the backbones of your duplicate helix." in my solution posted on the PyMOL mailing list.
- The last three commands utilize options (
The duplicated RNA helix is illustrated in the image below.
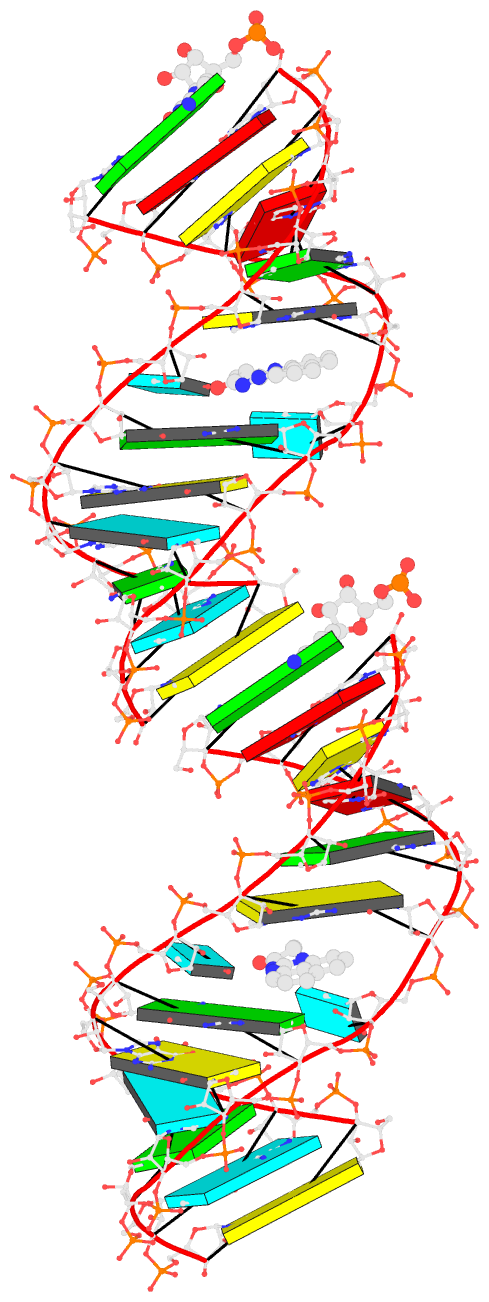
Some caveats
The original 13 base-pair RNA duplex (model.pdb) contains three main PDB format inconsistencies:
- Missing Chain Identifiers: The two strands lack proper chain identifiers in column 22.
- Incorrect Covalent Bond Distance: Nucleotides RU25 and RC26 are not covalently linked. Specifically, the distance between O3' of RU25 and P of RC26 is 3.5 Å, exceeding the expected 1.6 Å for a proper covalent bond.
- Misclassified Ligand Record: The ligand (LIG27) is incorrectly designated as
ATOMinstead of the appropriateHETATMrecord.