Announcing wDSSR: The Next-Generation Web Interface to X3DNA-DSSR
Dear 3DNA/DSSR Community,
We are thrilled to announce the official launch of wDSSR (https://web.x3dna-dssr.org/), the powerful new web interface to the X3DNA-DSSR analytical engine.
Developed by Drs. Shuxiang Li and Xiang-Jun Lu and supported by NIH grant R24GM153869, wDSSR represents a major leap forward from our highly popular 2019 Web 3DNA 2.0 framework. While Web 3DNA 2.0 has faithfully served the community for the analysis, visualization, and modeling of 3D nucleic acid structures, wDSSR was built from the ground up to take full advantage of modern web technologies and the latest DSSR backend capabilities.
A Modern, Streamlined Scientific Workflow
We have completely overhauled the user interface to provide a clean, intuitive, and task-driven experience. The core modeling and analysis tools are now seamlessly organized into a logical, single-word scientific workflow: Analyze, Rebuild, Model, Circularize, Mutate, Assemble, and Visualize.
Spotlight Feature: The "Assemble" Module
One of the most exciting upgrades is the newly renamed Assemble tab (formerly "Composite"). This advanced composite model builder allows you to effortlessly construct complex, higher-order models by linking any combination of nucleic acid duplexes or protein-DNA/RNA complexes. You can quickly connect up to six distinct target structures, ranging from simple linked A-DNA and B-DNA duplexes to large, protein-decorated structural assemblies.
Immediate Global Adoption
Although wDSSR has just launched, we are incredibly humbled to share that it is already seeing rapid worldwide adoption! According to recent network infrastructure data, the new interface is actively being used by researchers across North America, South America, Europe, and Asia. Within just a few days, we have recorded active sessions from prestigious institutions around the globe, including:
- The Weizmann Institute of Science in Israel
- Katholieke Universiteit Leuven in Belgium
- Queen's University in Canada
- Universidad Nacional Autonoma de Mexico (UNAM) in Mexico
- Emory University and the Wadsworth Centers Laboratories and Research in the United States
- Jawaharlal Nehru University and the China Education and Research Network in Asia
How to Cite
While a dedicated paper for wDSSR is currently in preparation, researchers should cite the server using its URL (https://web.x3dna-dssr.org/) alongside the 2019 Web 3DNA 2.0 paper and the foundational 2015 DSSR paper. Full details and funding acknowledgements can be found on our newly consolidated About page.
We invite you all to try out the new wDSSR platform! As always, your feedback is invaluable to us, and we encourage you to share your thoughts, questions, and structural models via the newly updated Questions & Feedback link in the wDSSR footer.
Happy modeling!
Any analysis of nucleic acid structures start with the identification of nucleotides (nts), the basic building unit. As per the PDB convention, each nt (like any other ligands) is specified by a three-letter identifier. For example, the four standard RNA nts are ..A, ..C, ..G, and ..U, respectively. The four corresponding standard DNA nts are .DA, .DC, .DG, and .DT, respectively. Note that here, for visualization purpose, each space is represented by a dot (.). In practice, the following codes for the five standard DNA/RNA nts — ADE, CYT, GUA, THY, and URA — are also commonly encountered, among other variants.
On top of the standard nts, there are numerous modified ones, each assigned a unique three-letter code. In the classic yeast phenylalanine tRNA (PDB id: 1ehz), 14 out of the 76 nts are modified, as shown in Fig. 1 below.

Fig. 1: Modified nucleotides in yeast phenylalanine tRNA 1ehz
It is challenging to maintain a comprehensive and updated list of ever-inceasing nts encountered in the PDB and molecular dynamics (MD) simulation packages (e.g., AMBER, GROMACS, and CHARMM). Thus, as of today, some well-known DNA/RNA structural bioinformatics tools can handle only standard nts or a limited list of modified ones.
From early on in the development of 3DNA, I observed that all recognized nts have a core six-membered ring, with atoms named N1,C2,N3,C4,C5,C6 consecutively (see Fig. 2 below). Purines have three additional atoms, named N7,C8,N9. So it is feasible to automatically identify nts, and classify them as pyrimidines and purines, based on the common core skeleton shared by all of them. Moreover, the ‘skeleton’ is not effected by any possible tautomeric or protonation state.

Fig. 2: Identification of nts in 3DNA/DSSR based on atomic names and planar geometry
Early versions of 3DNA employed only three atoms (N1, C2 and C6) and three distances to decide a nt. Purines were further discriminated by the N9 atom, and the N1–N9 distance. While developing DSSR, I revised the nt-identification algorithm by using a least-squares fitting procedure that makes use of all available base ring atoms instead of selected ones. The same new algorithm has also been adapted into the find_pair/analyze etc programs in 3DNA, as of v2.2.
As always, the idea can be best illustrated with a worked example. Guanine in its standard base reference frame, with the following list of nine ring atoms coordinates, is chosen for the least-squares fitting. See file Atomic_G.pdb in the 3DNA distribution, and also Table 1 of the report A Standard Reference Frame for the Description of Nucleic Acid Base-pair Geometry.
ATOM 2 N9 G A 1 -1.289 4.551 0.000
ATOM 3 C8 G A 1 0.023 4.962 0.000
ATOM 4 N7 G A 1 0.870 3.969 0.000
ATOM 5 C5 G A 1 0.071 2.833 0.000
ATOM 6 C6 G A 1 0.424 1.460 0.000
ATOM 8 N1 G A 1 -0.700 0.641 0.000
ATOM 9 C2 G A 1 -1.999 1.087 0.000
ATOM 11 N3 G A 1 -2.342 2.364 0.001
ATOM 12 C4 G A 1 -1.265 3.177 0.000
By using a ls-fitting procedure, only (any) three atoms are needed. We no longer need to make explicit selection, as we did previously (N1,C2,C6 and N9), thus allowing for possible modification on these atoms.
Using four nts (G1, 2MG10, H2U16, and PSU39, see Fig. 1 above top) of 1ehz as examples, the following list gives the atomic coordinates of base ring atoms, and root-mean-squres devisions (rmsd) of the least-squares fit. Of course, when performing least-squares fitting, the names of corresponding atoms must match (note the different ordering of atoms for H2U and PSU in the list vs the above standard G reference).
#G1, rmsd=0.008
ATOM 14 N9 G A 1 51.628 45.992 53.798 1.00 93.67 N
ATOM 15 C8 G A 1 51.064 46.007 52.547 1.00 92.60 C
ATOM 16 N7 G A 1 51.379 44.966 51.831 1.00 91.19 N
ATOM 17 C5 G A 1 52.197 44.218 52.658 1.00 91.47 C
ATOM 18 C6 G A 1 52.848 42.992 52.425 1.00 90.68 C
ATOM 20 N1 G A 1 53.588 42.588 53.534 1.00 90.71 N
ATOM 21 C2 G A 1 53.685 43.282 54.716 1.00 91.21 C
ATOM 23 N3 G A 1 53.077 44.429 54.946 1.00 91.92 N
ATOM 24 C4 G A 1 52.356 44.836 53.879 1.00 92.62 C
#2MG10, rmsd=0.018
HETATM 207 N9 2MG A 10 61.581 47.402 18.752 1.00 42.14 N
HETATM 208 C8 2MG A 10 62.199 48.621 18.635 1.00 40.38 C
HETATM 209 N7 2MG A 10 63.494 48.534 18.422 1.00 40.70 N
HETATM 210 C5 2MG A 10 63.745 47.167 18.395 1.00 43.82 C
HETATM 211 C6 2MG A 10 64.965 46.449 18.205 1.00 43.45 C
HETATM 213 N1 2MG A 10 64.767 45.086 18.293 1.00 44.71 N
HETATM 214 C2 2MG A 10 63.541 44.482 18.486 1.00 47.21 C
HETATM 217 N3 2MG A 10 62.411 45.125 18.614 1.00 45.85 N
HETATM 218 C4 2MG A 10 62.574 46.451 18.582 1.00 43.27 C
#H2U16, rmsd=0.188
HETATM 336 N1 H2U A 16 77.347 53.323 34.582 1.00 91.19 N
HETATM 337 C2 H2U A 16 76.119 52.865 34.160 1.00 92.39 C
HETATM 339 N3 H2U A 16 75.123 52.894 35.107 1.00 93.28 N
HETATM 340 C4 H2U A 16 75.289 52.711 36.458 1.00 93.34 C
HETATM 342 C5 H2U A 16 76.696 52.479 36.909 1.00 93.77 C
HETATM 343 C6 H2U A 16 77.717 53.238 36.039 1.00 93.22 C
#PSU39, rmsd=0.004
HETATM 845 N1 PSU A 39 74.080 36.066 5.459 1.00 75.82 N
HETATM 846 C2 PSU A 39 74.415 36.835 4.354 1.00 75.59 C
HETATM 847 N3 PSU A 39 75.735 36.769 3.984 1.00 76.29 N
HETATM 848 C4 PSU A 39 76.728 36.038 4.591 1.00 77.28 C
HETATM 849 C5 PSU A 39 76.307 35.280 5.732 1.00 77.93 C
HETATM 850 C6 PSU A 39 75.025 35.316 6.112 1.00 76.07 C
As noted in the DSSR paper, the rmsd is normally <0.1 Å since base rings are rigid. To account for experimental error and special non-planar cases, such as H2U in 1ehz, the default rmsd cutoff is set to 0.28 Å by default.
With the above detailed algorithm, DSSR (and the 3DNA find_pair/analyze programs) can automatically identify virtually all ‘recognizable’ nts in the PDB. A survey performed in June 2015 detected 630 different types of modified nucleotides in the PDB.
It is worth noting the following points:
- The choice of standard G instead of A as the reference base has no impact on the results. As a matter of fact, the rmsd between G and A is only 0.04 Å. Note also the generous default cutoff of 0.28 Å.
- The method obviously depends on proper naming of the ring atoms. Specially, the base ring atoms must be named
N1,C2,N3,C4,C5,C6 consecutively, with purines having three additional atoms named N7,C8,N9. Thus, under this scheme, TPP (thiamine diphosphate) would not be recognized as a nt by default, simply because of the extra prime (′) of atoms in the six-membered ring. In nucleic acid structures, the prime symbol is normally associated with atoms of the sugar moiety (e.g., the C5′ atom).
")
Fig. 3: TPP (thiamine diphosphate) would not be recognized as a nt.
- On the other hand, nt cofactors in an otherwise ‘pure’ protein structure will also be recognized. One example is the two AMP (adenosine monophosphate) ligands in PDB entry 12as. This extra identification of nts does no harm in such cases. As shown in the analysis of the SAM-I riboswitch in the DSSR paper, taking the SAM ligand as a nt in base triplet recognition is a neat feature.
- Once a nucleotide has been identified and classified into purines and pyrimidines, exocyclic atoms can be used for further assignment:
O6 or N2 distinguishes guanine from adenine, N4 separates cytosine from thymine and uracil, and C7 (or C5M, the methyl group) differentiates thymine from uracil. For some modified nts, the distinctions within purines or pyrimidines may not be that obvious. For example, inosine may be taken as a modified guanine or adenine. However, this ambiguity does not pose any significant effect on the calculated base-pair parameters.
- In DSSR and 3DNA, each identified nt is assigned a one-letter shorthand code: the standard
..A, .DA, and ADE (among a few other common variations) is shortened to upper-case A, and similarly for C, G, T, and U. Modified nts, on the other hand, are shortened to their corresponding lower-case symbol. For example, modified guanine such as 2MG and M2G in the yeast phenylalanine tRNA (see Fig. 1 above) is assigned g. So in 3DNA/DSSR output, the upper and lower cases of bases (e.g., nts=3 gCG A.2MG10,A.C25,A.G45) convey special meanings.
Related topics:

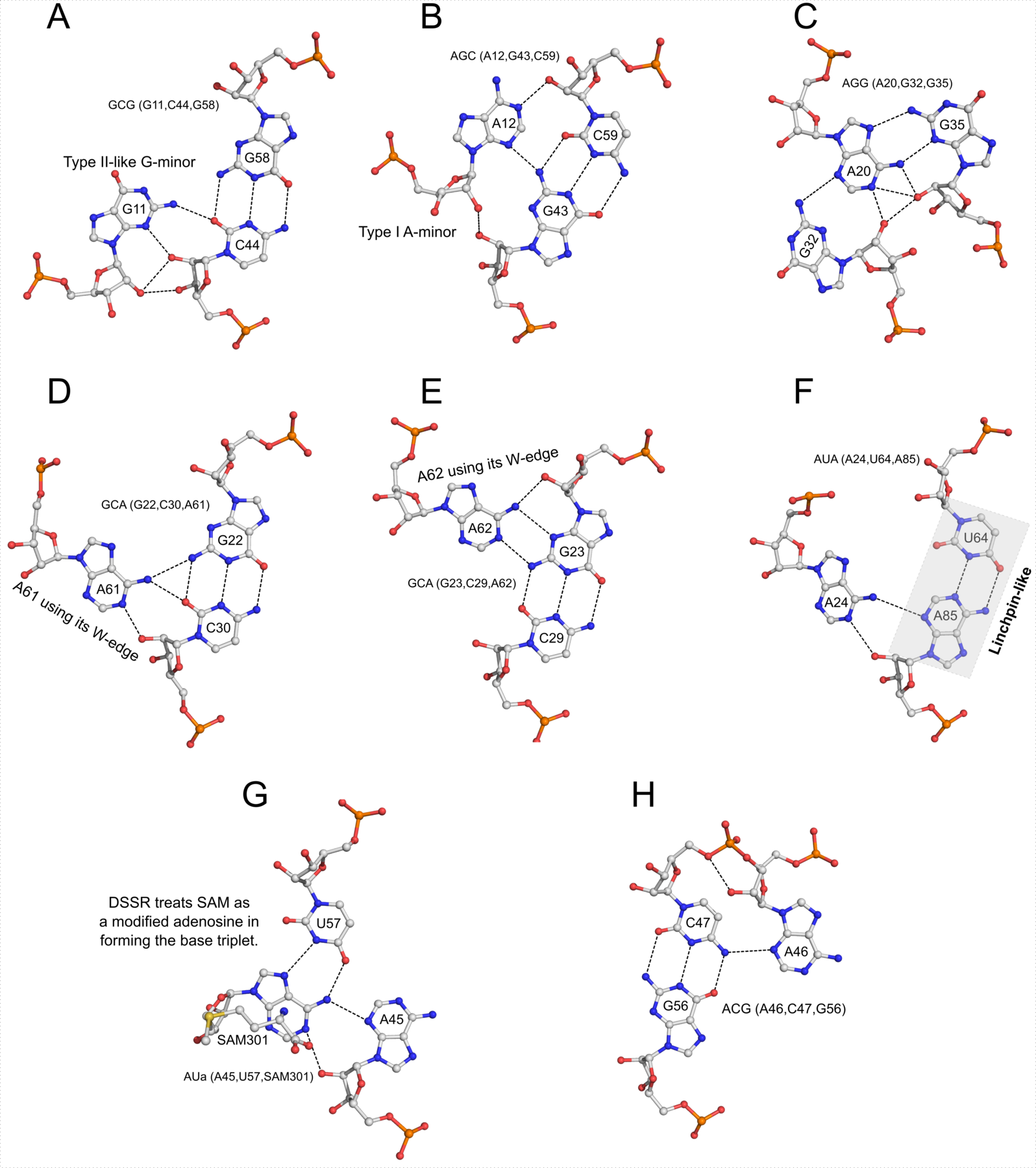
In DSSR (and find_pair -p from the original 3DNA suite), multiplets is defined as “three or more bases associated in a coplanar geometry via a network of hydrogen-bonding interactions. Multiplets are identified through inter-connected base pairs, filtered by pair-wise stacking interactions and vertical separations to ensure overall coplanarity.”
DSSR detects multiplets automatically, and outputs a corresponding MODEL/ENDMDL delineated PDB file (dssr-multiplets.pdb by default) where each multiplet is laid in the most extended view in terms of base planes. The DSSR Nucleic Acids Research (NAR) paper contains four examples (in supplemental Figures 1, 3, 4, and 7) to illustrate this functionality. Please refer to Reproducing results published in the DSSR-NAR paper on the 3DNA Forum for details.
Recently, I read the article titled InterRNA: a database of base interactions in RNA structures by Appasamy et al. in NAR. In Figure 2 of the paper, the authors showcased a sextuple (hexaplet) identified in the E. coli ribosome (PDB id: 4tpe), along with six base-base H-bonds contained therein.
With interest, I tried to run DSSR on the PDB entry 4tpe. As it turns out, ‘4tpe’ has been merged into 4u27 in mmCIF format. I ran DSSR (v1.4.6-2015dec16) in its default settings on ‘4u27’ and get the following summary of results.
# x3dna-dssr -i=4u27.cif -o=4u27.out
total number of base pairs: 4822
total number of multiplets: 680
total number of helices: 264
total number of stems: 566
total number of isolated WC/wobble pairs: 193
total number of atom-base capping interactions: 615
total number of hairpin loops: 215
total number of bulges: 137
total number of internal loops: 244
total number of junctions: 108
total number of non-loop single-stranded segments: 83
total number of kissing loops: 14
total number of A-minor (type I and II) motifs: 246
total number of ribose zippers: 127
total number of kink turns: 15
Among the 680 DSSR-identified multiplets, two hexaplets (one on chain “AA”, and another on “CA”) match those reported by Appasamy et al., as shown below:
678 nts=6 GUUAAA 1:AA.G404,1:AA.U438,1:AA.U439,1:AA.A496,1:AA.A498,1:AA.A499
679 nts=6 GUUAAA 1:CA.G404,1:CA.U438,1:CA.U439,1:CA.A496,1:CA.A498,1:CA.A499
For illustration, the hexaplet #678 is extracted from dssr-multiplets.pdb to file 4u27-hexaplet.pdb (download the coordinates) and shown below. The figure is generated by DSSR and PyMOL, as detailed in Reproducing results published in the DSSR-NAR paper on the 3DNA Forum.
x3dna-dssr -i=4u27-hexaplet.pdb -o=4u27-hexaplet.pml --hbfile-pymol

DSSR-identified hexaplet GUUAAA in 4u27.
DSSR identifies 6 base pairs in the hexaplet:
# x3dna-dssr -i=4u27-hexaplet.pdb --idstr=short
List of 6 base pairs
nt1 nt2 bp name Saenger LW DSSR
1 G404 A498 G+A -- n/a tSS tm+m
2 G404 A499 G+A -- n/a cWH cW+M
3 U438 A496 U-A rHoogsteen 24-XXIV tWH tW-M
4 U439 A496 U-A -- n/a cH. cM-.
5 U439 A498 U-A WC 20-XX cWW cW-W
6 A496 A498 A+A -- n/a cWH cW+M
It detects a total of 9 H-bonds as shown below. In addition to the 6 base-base H-bonds noted by Appasamy et al., DSSR also finds 3 sugar-base H-bonds (#1, #2, and #4, labeled in green) that obviously play a role in stabilizing the high-order base association.
# x3dna-dssr -i=4u27-hexaplet.pdb --get-hbonds --idstr=short
11 59 #1 o 3.017 O:N O2'@G404 N3@U439
11 104 #2 o 2.578 O:N O2'@G404 N1@A498
18 125 #3 p 3.089 O:N O6@G404 N6@A499
21 96 #4 o 3.289 N:O N2@G404 O2'@A498
21 106 #5 p 2.797 N:N N2@G404 N3@A498
39 78 #6 p 2.944 N:N N3@U438 N7@A496
61 81 #7 p 3.167 O:N O4@U439 N6@A496
61 103 #8 p 2.662 O:N O4@U439 N6@A498
82 103 #9 p 3.152 N:N N1@A496 N6@A498
From the Jmol mailing list, I noticed Jmol 14.4.0 was released yesterday (October 13, 2015) by Dr. Bob Hanson. Among the development highlights is the following item:
biomolecule annotations including DSSR, RNA3D, EBI sequence domains, and PDB validation data
I am glad to see that DSSR has been integrated into Jmol, one of the most popular molecular graphics visualization programs. To enable easy access to the DSSR functionality from Jmol, I’ve set up two websites with easy-to-remember URLs: http://jmol.x3dna.org and http://jsmol.x3dna.org. They both point to the same jsmol/ folder extracted from jsmol.zip of the Jmol distribution.
In retrospect, I first met Bob at the Workshop on the PDBx/mmCIF Data Exchange Format for Structural Biology held at Rutgers University during October 21-22, 2013. I approached him during a lunch break, asking for a possible collaboration on integrating DSSR into Jmol. The name DSSR may have played a role in convincing Bob, since it matches the well-known DSSP program for proteins. In the end, we were both excited about the project, talked into details after the meeting, and continued our conversation the next morning while I drove him to the airport.
Nothing real happened until early April 2014. Once getting started, however, we moved forward rapidly: it took less then three weeks to get the first functional version ready for the community to play. See Bob’s announcement RNA/DNA Secondary Structure, anyone? in the Jmol mailing list on April 9, 2014. During this process, we communicated extensively via email, up to 30 messages per day, on technical details for better communication between the two programs. The integration works by using Jmol as a front-end, which calls a web-serivce hosted at Columbia University for DSSR analysis. Jmol’s parsing of the DSSR output is facilitated by the dedicated --jmol option.
The above preliminary, yet functional, DSSR-Jmol integration had be in service without infrastructural changes until two months ago. In August 10, 2015, Bob contacted me:
I might make a significant request though. That would be for the server to deliver all this in JSON format. This is really the way to go. It is what people want and it is perfect for Jmol as well.
I’d played around with JSON or SQLite as a structured data exchange format for quite some time, and Bob’s request finally convinced me that JSON is the (better) way to go. And that began another around of intensive collaborative work that has switched the exchange format between DSSR and Jmol from plain text output to JSON. From August 10 to September 22, we had a total of over 170-email exchanges, plus Skype. JSON has really simplified lives of both parties, especially in the long run.
Overall, collaborating with Bob has been truly an enjoyable and rewarding experience. The DSSR-Jmol integration also serves as a concrete example of what can be achieved by two dedicated minds with complementary expertise.
Over the past couple of weeks, I’ve added two more DSSR options, --symmetry and --nmr, that are closely related to an ensemble of MODEL/ENDMDL-delineated structures in PDB files. However, there exist subtle differences between the two cases, and the usage of the same MODEL/ENDMDL ensemble format can be ambiguous to the uninitiated. This blog post aims to clarify the issues, using concrete examples.
The --symmetry options applies to X-ray crystal structures where an asymmetric unit represents only part of the whole biological assembly. In standard PDB format, the asymmetric unit contains instructions to produce crystallographic symmetry
related molecules.. Nevertheless, the biological assembly are also provided by the PDB (or NDB), with coordinate files ending with .pdb1 or such. For example, the PDB entry 2d94 has the single-stranded sequence GGGCGCCC in its asymmetric unit (2d94.pdb). It is the biological assembly in file 2d94.pdb1 that contains the DNA double helix.
x3dna-dssr -i=2d94.pdb # no pairs found
x3dna-dssr -i=2d94.pdb1 # still no pairs found
x3dna-dssr -i=2d94.pdb1 --symm # 8 pairs found
x3dna-dssr -i=2d94.pdb --symm # no pairs found
As shown by the above examples, DSSR by default reads only the first model even given the biological assemble file 2d94.pdb1. It is with --symmetry (abbreviated to --symm) explicitly specified that DSSR takes all models in the input biological assemble file into consideration. The last case also illustrates that DSSR does not generate crystallographic symmetry related molecules. The --symm simply informs DSSR to take all models, which already exist in the input file, into consideration.
On the other hand, the --nmr option is for auto-processing an ensemble of structures solved by solution NMR method (or trajectories of molecular dynamics simulations). The key point here is that each of the MODEL/ENDMDL-delinated structures is independent and thus can be processed separately, even though they are obviously closely related. Using the PDB entry 2n2d as an example, here are some sample usages:
x3dna-dssr -i=2n2d.pdb -o= 2n2d-first.out # only the first structure is processed
x3dna-dssr -i=2n2d.pdb --nmr -o=2n2d-all.out # all 10 structures are processed
x3dna-dssr -i=2n2d.pdb --nmr --json -o=2n2d-all.json # ibid., with output in JSON
Note that the NMR file is named 2n2d.pdb, and it contains 10 structures.
Interesting mixes show up when an X-ray biological assembly with multiple MODEL/ENDMDL entries is analyzed with --nmr, or an NMR entry is handled with --symmetry. Here are two such examples:
x3dna-dssr -i=2d94.pdb1 --nmr -o=temp # models 1 and 2 are handled sepatately
x3dna-dssr -i=2n2d.pdb --symm -o=temp # wrong -- does not make sense!
In summary, the --symmetry option is intended to treat symmetry-related molecules as a whole, as in a biological assembly of X-ray crystal structures. In contrast, the --nmr option aims to automate the analysis of each structure in a MODEL/ENDMDL-delineated ensemble, as in NMR structures or trajectories of MD simulations. The distinction between the two MODEL/ENDMDL usages is most clearly seen via a molecular visualization program: for example, check the figure below for 2d94.pdb1 (left) and 2n2d.pdb (right) when all frames are selected using Jmol.
Recently, I read with great interest an article titled A context-sensitive guide to RNA & DNA base-pair & base-stack geometry by Dr. Jane Richardson, published in CCN (Computational Crystallography Newsletter, 2015, 5, 42—49). Highlighted in the article are Buckle and Propeller twist (see bottom left of the figure below), two of the angular parameters that characterize base-pair (bp) non-planarity. Particularly, I was intrigued by the “Notes on measures and figures” at the end:
Base normals were constructed in Mage (Richardson 2001) and twist torsions and buckle angles were measured from them; propeller-twists were measured as dihedral angles around an axis between N1/9 atoms.

The Richardson CCN article prompted me to think more on intuitive description of bp geometry that can be easily grasped by experimentalist, especially X-ray crystallographers or cryo-EM practitioners. Without worrying about model building as with the six rigid-body parameters, it is straightforward to come up with a new set of four ‘simple’ parameters (Shear, Stretch, Buckle and Propeller) with the following characteristics:
- Each parameter can be positive or negative. For type M–N pairs (as in the canonical cases), Shear and Buckle reverse their signs when the two bases are swapped (i.e. counted as N–M instead of M–N). In all other cases, the signs of the parameters remain unchanged. See the DSSR paper for the definition of M+N vs M–N type of pairs.
- Intuitive results for non-canonical pairs, even when Opening is ~180º.
- Consistent definition between Shear/Buckle (_x_-axis) vs Stretch/Propeller (_y_-axis).
- As in 3DNA and DSSR, Buckle^2 + Propeller^2 = interBase_angle^2. Either Buckle or Propeller can render the two base planes of a pair non-parallel. Combined together, they introduce a non-zero inter-base angle. By definition, each parameter should not be larger than the overall inter-base angle.
With the cartoon-block representation introduced in DSSR, base-stacking interactions and bp deformations (especially Buckle and Propeller) are immediately obvious. Two example are illustrated in the figure below: one is the classic Dickerson B-DNA dodecamer (355d, DSSR output), and the other is the parallel double-stranded helix of poly(A) RNA (4jrd, DSSR output).
A portion of DSSR output for the B-DNA duplex 355d is shown below. Note that the first bp (at the bottom left in the figure above) has a Propeller of –17º (and a Buckle of +7º). As beautifully explained by Calladine et al. in their book Understanding DNA,
The Molecule & How It Works, Watson-Crick pairs prefer to have negative Propeller in right-handed DNA double helices to improve same-strand base-stacking interactions. The average value of Propeller in A- and B-DNA crystal structures is around –11º (see Table 3 of the Olson et al. standard base reference frame paper).
nt1 nt2 bp name Saenger LW DSSR
1 A.DC1 B.DG24 C-G WC 19-XIX cWW cW-W
[-105.9(anti) ~C2'-endo lambda=53.5] [-141.3(anti) ~C3'-endo lambda=52.7]
d(C1'-C1')=10.71 d(N1-N9)=8.96 d(C6-C8)=9.88 tor(C1'-N1-N9-C1')=-21.4
H-bonds[3]: "O2(carbonyl)-N2(amino)[2.83],N3-N1(imino)[2.90],N4(amino)-O6(carbonyl)[2.98]"
interBase-angle=19 Simple-bpParams: Shear=0.28 Stretch=-0.13 Buckle=7.3 Propeller=-17.2
bp-pars: [0.28 -0.14 0.07 6.93 -17.31 -0.61]
2 A.DG2 B.DC23 G-C WC 19-XIX cWW cW-W
[-85.4(anti) ~C2'-endo lambda=53.4] [-150.3(anti) ~C3'-endo lambda=55.4]
d(C1'-C1')=10.61 d(N1-N9)=8.92 d(C6-C8)=9.83 tor(C1'-N1-N9-C1')=-21.7
H-bonds[3]: "O6(carbonyl)-N4(amino)[2.91],N1(imino)-N3[2.88],N2(amino)-O2(carbonyl)[2.88]"
interBase-angle=17 Simple-bpParams: Shear=-0.24 Stretch=-0.18 Buckle=9.0 Propeller=-14.5
bp-pars: [-0.24 -0.18 0.49 9.34 -14.30 -2.08]
A portion of DSSR output for the parallel A-DNA duplex 4jrd is shown below. Note that the values of ‘simple’ Propeller are positive for both bps #7 and #8. In contrast, the rigid-body bp parameters have their signs flipped over when Opening is switched from –179.56º for bp#7 to +179.23º for bp#8. This sign ‘ambiguity’ around 180º Opening could be confusing. Yet, all the six bp parameters must be kept as they are for rigorous rebuilding, especially within a larger context than a bp per se. From the very beginning, 3DNA has adopted the convention of keeping angular parameters in the range of [–180º, +180º] instead of [0, 360º], allowing left-handed Z-DNA to have negative twist.
7 A.A8 B.A7 A+A -- 02-II tHH tM+M
[-175.8(anti) ~C3'-endo lambda=10.2] [-172.7(anti) ~C3'-endo lambda=12.6]
d(C1'-C1')=11.15 d(N1-N9)=8.29 d(C6-C8)=6.31 tor(C1'-N1-N9-C1')=160.1
H-bonds[4]: "OP2-N6(amino)[2.97],N7-N6(amino)[2.97],N6(amino)-OP2[2.92],N6(amino)-N7[2.91]"
interBase-angle=14 Simple-bpParams: Shear=-7.88 Stretch=0.66 Buckle=-7.8 Propeller=11.9
bp-pars: [-6.00 5.15 -0.02 0.63 14.22 -179.56]
8 A.A9 B.A8 A+A -- 02-II tHH tM+M
[-177.4(anti) ~C3'-endo lambda=12.4] [-175.8(anti) ~C3'-endo lambda=10.3]
d(C1'-C1')=11.01 d(N1-N9)=8.15 d(C6-C8)=6.18 tor(C1'-N1-N9-C1')=158.5
H-bonds[4]: "OP2-N6(amino)[2.93],N7-N6(amino)[2.88],N6(amino)-OP2[2.97],N6(amino)-N7[2.92]"
interBase-angle=15 Simple-bpParams: Shear=-7.91 Stretch=0.56 Buckle=-7.0 Propeller=13.7
bp-pars: [6.11 -5.06 -0.05 -2.26 -15.22 179.23]
Standard nitrogenous bases in DNA and RNA (A, C, G, T, and U) are aromatic compounds, each with a planar geometry. In the analyses of three-dimensional (3D) nucleic acid structures, the planar bases are normally taken as rigid bodies. The relative geometry of the two bases in base pair (bp) can then be rigorously quantified by six rigid-body parameters (see figure below). The three translations along the _x_-, _y_- and _z_-axes are termed Shear, Stretch, and Stagger, respectively. The three corresponding rotations are called Buckle, Propeller (twist), and Opening.
3DNA is unique with its coupled analyze and rebuild programs. The former calculates six bp parameters given 3D atomic coordinates (in PDB or PDBx/mmCIF format), while the later takes a set of such parameters to generate the corresponding structure. The rigor of the description can be easily verified in two equivalent ways: the close to zero root-mean-square deviation (RMSD) between the rebuilt structure and the original coordinates, after a least-squares superposition; or the identical six bp parameters when the rebuilt structure is analyzed.
As is often the case, a concrete example would make the point clear. Here I am using the reverse Hoogsteen (rHoogsteen) bp between U8 and A14 (see image below) in the yeast phenylalanine tRNA (1ehz) as an example. The PDB atomic coordinates of the U8–A14 rHoogsteen pair, excluding backbone atoms except for C1′, is stored in file 1ehz-U8-A14.pdb.
")
find_pair 1ehz-U8-A14.pdb stdout | analyze stdin
# bp parameters in file '1ehz-U8-A14.out'
# also generated 'bp_step.par' for rebuilding below
rebuild -atomic bp_step.par 1ehz-U8-A14-3DNA.pdb
# rmsd is 0.044 Å between '1ehz-U8-A14.pdb' and '1ehz-U8-A14-3DNA.pdb'
find_pair 1ehz-U8-A14-3DNA.pdb stdout | analyze stdin
# bp parameters of the rebuilt structure in '1ehz-U8-A14-3DNA.out'
rebuild -atomic bp_step.par 1ehz-U8-A14-3DNA-new.pdb
# rmsd is 0 Å between '1ehz-U8-A14-3DNA.pdb' and '1ehz-U8-A14-3DNA-new.pdb'
Note that the above commands should be performed in order, since the file bp_step.par is overwritten after each analyze run. For your verification, here are the links to the five files:
The 0.044 Å rmsd between the original PDB coordinates in 1ehz-U8-A14.pdb and the 3DNA rebuilt structure in 1ehz-U8-A14-3DNA.pdb is due to the slight non-planarity of experimental bases. The rmsd is 0 between the two rounds of 3DNA rebuilt structures, 1ehz-U8-A14-3DNA.pdb and 1ehz-U8-A14-3DNA-new.pdb, as expected.
The bp parameters in 1ehz-U8-A14.out and 1ehz-U8-A14-3DNA.out are identical, as expected, and they are shown below.
Local base-pair parameters
bp Shear Stretch Stagger Buckle Propeller Opening
1 U-A 4.14 -1.91 0.77 -4.62 12.12 -103.09
Running DSSR on 1ehz-U8-A14.pdb gives the following results. Note that the six bp parameters (last row prefixed with bp-pars) are the exactly same as in 3DNA — we are consistent.
# x3dna-dssr -i=1ehz-U8-A14.pdb --more
List of 1 base pair
nt1 nt2 bp name Saenger LW DSSR
1 A.U8 A.A14 U-A rHoogsteen 24-XXIV tWH tW-M
[n/a(n/a) ---- lambda=28.3] [n/a(n/a) ---- lambda=21.5]
d(C1'-C1')=9.63 d(N1-N9)=7.06 d(C6-C8)=6.00 tor(C1'-N1-N9-C1')=174.4
H-bonds[2]: "O2(carbonyl)-N6(amino)[3.00],N3(imino)-N7[2.74]"
interBase-angle=12.97 Simple-bpParams: Shear=4.28 Stretch=1.55 Buckle=-11.8 Propeller=5.4
bp-pars: [4.14 -1.91 0.77 -4.62 12.12 -103.09]
As mentioned in the recent DSSR paper:
As in 3DNA (6,7), DSSR takes advantage of the six standard base-pair parameters––three translations (Shear, Stretch, Stagger) and three rotations (Buckle, Propeller, Opening)––to quantify the relative spatial position and orientation of any two interacting bases rigorously. Among the six parameters, only Shear, Stretch, and Opening are critical for characterizing different types of pairs. Buckle, Propeller and Stagger, on the other hand, describe the nonplanarity of a given pair (6). By virtue of the definition of the standard base reference frame, Shear, Stretch, and Opening are all close to zero for Watson-Crick pairs. Moreover, every other type of pair has a set of characteristic parameters. For example, the wobble G–U pair is characterized by an average Shear of –2.2 Å, and the Hoogsteen A+U pair is distinguished by a Stretch of approximately –3.5 Å and an Opening of near 66º.
In a follow-up post, I will talk about the “simple” bp parameters (Simple-bpParams in the above DSSR output list) recently introduced into DSSR — stay tuned!
As of DSSR v1.3.0-2015aug27, the --json option is available for producing analysis results that is strictly compliant with the JSON data exchange format. The JSON file contains numerous DSSR-derived structural features, including those in the default main output, backbone torsions in dssr-torsions.txt, and a detailed list of hydrogen bonds.
According to the official JSON website:
JSON (JavaScript Object Notation) is a lightweight data-interchange format. It is easy for humans to read and write. It is easy for machines to parse and generate. It is based on a subset of the JavaScript Programming Language… JSON is a text format that is completely language independent… These properties make JSON an ideal data-interchange language.
Indeed, the JSON output file makes DSSR readily accessible for integration with other bioinformatics tools or normal usages from the command line. Using the classic yeast phenylalanine tRNA 1ehz as an example (1ehz.pdb), let’s go over some simple use-cases. Note that the following examples take advantage of jq, a lightweight and flexible command-line JSON processor.
x3dna-dssr -i=1ehz.pdb --json -o=1ehz-dssr.json
jq . 1ehz-dssr.json # reformatted for pretty output
x3dna-dssr -i=1ehz.pdb --json | jq . # the above 2 steps combined
With 1ehz-dssr.json in hand, we can easily extract DSSR-derived structural features of interest:
jq .pairs 1ehz-dssr.json # list of 34 pairs
jq .multiplets 1ehz-dssr.json # list of 4 base triplets
jq .hbonds 1ehz-dssr.json # list of hydrogen bonds
jq .helices 1ehz-dssr.json
jq .stems 1ehz-dssr.json
# list of nucleotide parameters, including torsion angles and suites
jq .ntParams 1ehz-dssr.json
# list of 14 modified nucleotides
jq '.ntParams[] | select(.is_modified)' 1ehz-dssr.json
# select nucleotide id, delta torsion, sugar puckering and cluster of suite name
jq '.ntParams[] | {nt_id, delta, puckering, cluster}' 1ehz-dssr.json
# same selection as above, but in 'Comma Separated Values' format
jq -r '.ntParams[] | [.nt_id, .delta, .puckering, .cluster] | @csv' 1ehz-dssr.json
Here is the result of running jq (v1.5) to select multiplets:
# jq .multiplets 1ehz-dssr.json
[
{
"index": 1,
"num_nts": 3,
"nts_short": "UAA",
"nts_long": "A.U8,A.A14,A.A21"
},
{
"index": 2,
"num_nts": 3,
"nts_short": "AUA",
"nts_long": "A.A9,A.U12,A.A23"
},
{
"index": 3,
"num_nts": 3,
"nts_short": "gCG",
"nts_long": "A.2MG10,A.C25,A.G45"
},
{
"index": 4,
"num_nts": 3,
"nts_short": "CGg",
"nts_long": "A.C13,A.G22,A.7MG46"
}
]
With the JSON file, DSSR can now be connected with the bioinformatics community in a ‘structured’ way, with a clearly delineated boundary. Now I can enjoy the freedom of refining the default main output format, without worrying too much about breaking third-party parsers. Moreover, I no longer need to write an adapter for each integration of DSSR with other tools. So nice!
For your reference, here is the output file 1ehz-dssr.json. It may be possible that the identifiers (names) of the JSON output will be refined in the next few iterations. I welcome your comments to make the DSSR-derived JSON better suite your needs.
It is a great pleasure to note that a paper titled DSSR, an integrated software tool for dissecting the spatial structure of RNA has recently been published in Nucleic Acids Research (NAR). Co-authored by Harmen Bussemaker, Wilma Olson and me (a team with a unique combination of complementary expertise), this DSSR paper represents another solid piece of work that I feel proud of. In contrast to our previous GpU dinucleotide platform paper focusing on results, and the two major 3DNA papers concentrating on methods, the current NAR article describes significant scientific findings that are enabled by the novel analysis algorithms implemented in the program. Moreover, DSSR introduces an appealing and highly informative “cartoon-block” representation of RNA structures that combines PyMOL cartoon schematics with 3DNA base color-coded rectangular blocks.
The abstract of the paper is quoted below:
Insight into the three-dimensional architecture of RNA is essential for understanding its cellular functions. However, even the classic transfer RNA structure contains features that are overlooked by existing bioinformatics tools. Here we present DSSR (Dissecting the Spatial Structure of RNA), an integrated and automated tool for analyzing and annotating RNA tertiary structures. The software identifies canonical and noncanonical base pairs, including those with modified nucleotides, in any tautomeric or protonation state. DSSR detects higher-order coplanar base associations, termed multiplets. It finds arrays of stacked pairs, classifies them by base-pair identity and backbone connectivity, and distinguishes a stem of covalently connected canonical pairs from a helix of stacked pairs of arbitrary type/linkage. DSSR identifies coaxial stacking of multiple stems within a single helix and lists isolated canonical pairs that lie outside of a stem. The program characterizes ‘closed’ loops of various types (hairpin, bulge, internal, and junction loops) and pseudoknots of arbitrary complexity. Notably, DSSR employs isolated pairs and the ends of stems, whether pseudoknotted or not, to define junction loops. This new, inclusive definition provides a novel perspective on the spatial organization of RNA. Tests on all nucleic acid structures in the Protein Data Bank confirm the efficiency and robustness of the software, and applications to representative RNA molecules illustrate its unique features. DSSR and related materials are freely available at http://x3dna.org/.
During the review process, we are delighted that the referees confirmed the claim that we made in the cover letter: “We would also like to emphasize that our reported results are easily verifiable, and we assure rigorous reproducibility of the data and figures described in this article.” Now that the paper has been published, as a follow-up, I’ve made available all the scripts and data files associated with the paper in a new section DSSR-NAR paper on the 3DNA Forum. The DSSR User Manual has also been updated with additional, previously undocumented, auxiliary options.
Overall, it took me more than ten days to create the 19 posts in the DSSR-NAR paper section and to revise the DSSR User Manual, along with other minor refinements for consistency. During the process, I’ve tried to make the scripts and data files self-contained for wide accessibility and easy understanding.
Any interested party should now be able to reproduce the table and figures (including the supplementary data) reported in the article. Moreover, with the additional details given in the post RNA cartoon-block representations with PyMOL and DSSR, one can easily generate similar schematic images as shown below:
I feel confident to claim that the results reported in our DSSR paper are reproducible. If you have issues related to the paper, please post them on the 3DNA Forum. I strive to respond promptly to any questions asked there.
In summary, DSSR is an integrated computational tool, designed from the bottom up to streamline the analysis of RNA three-dimensional structures. It is built upon my extensive experience in supporting 3DNA, growing knowledge of RNA structures, and refined programming skills. DSSR has a combined set of functionalities well beyond the scope of any known specialized resources. The program may well serve as a cornerstone for RNA structural bioinformatics and will benefit a broad range of possible applications.
Nowadays, “big data” and “big science” are hot topics. They all sound good and certainly come about for a reason. Yet, to transform data to information to knowledge to understanding to wisdom, sophisticated software tools are required. The programs can be big and complicated, or small and self-contained, fitting different purposes. As long as they can get the claimed job done in a robust fashion, size should not be a concern.
Over the years, however, I have seen a trend of bloated software with many (fragile) dependencies in bioinformatics. Some tools are so picky and hard to use/maintain that instead of serving, they become sort of a master. As a more representative example, I recently tried to install an open-source software associated with a paper published just a few years ago in a leading journal. The software has only a few dependencies, yet some of them have already become obsolete. I spent hours each time, on Mac OS X and two versions of Ubuntu Linux, but failed to get it running properly (always abort with error messages). The download page hosting the software has been inactive since around the publication of the paper. Presumably, the PhD student or postdoc who wrote the code had left the lab, and with a paper published, all is done!
As an active practitioner of bioinformatics for well over a decade, I can confidently claim to be well above average in familiarity with Linux/Mac OS X and associated shell programming and make etc tools, and various common scripting and compiled programming languages. Yet, once in a while, I get frustrated when I try to download and install a software tool attached to a paper I am interested in. As I see it, the vast majority of software programs from research labs are publication-oriented — as long a paper is published, it is finished.
From my experience, I always see software as engineering. It needs careful design and great attention to meticulous details. A sophisticated piece of scientific software is a combination of science and engineering. Expertise in domain knowledge is a must, and refined skills in computer programming is indispensable. The DSSR program I created and continuously refined over the past three years represents what a scientific software should be in my believe.
Among other unique features, DSSR is tiny (< 1mb), self-contained (without run-time dependencies) and runs on Windows, Mac OS X, and Linux. Getting DSSR up and running should take only minutes by any one with basic familiarity of common computer systems. I have no doubt that the beauty of being small as represented by DSSR will be gradually appreciated by the community.
")
")
 in cartoon-block representation")
 RNA (4jrd)")
 with base blocks")
 with WC base-pair blocks")
 of the C-G pair that closes the GUAA tetraloop facing the viewer")
 -- base blocks in outline")
{kind=link}