 A video overview of DSSR
A video overview of DSSR
DSSR (Dissecting the Spatial Structure of RNA) is an integrated software tool for the analysis/annotation, model building, and schematic visualization of 3D nucleic acid structures (see the figures below and the video overview). It is built upon the well-known, tested, and trusted 3DNA suite of programs. DSSR has been made possible by the developer’s extensive user-support experience, detail-oriented software engineering skills, and expert domain knowledge accumulated over two decades. It streamlines tasks in RNA/DNA structural bioinformatics, and outperforms its ‘competitors’ by far in terms of functionality, usability, and support.
Wide citations. DSSR has been widely cited in scientific literature, including: (i) “Selective small-molecule inhibition of an RNA structural element” (Nature, 2015; Merck Research Laboratories), (ii) “The structure of the yeast mitochondrial ribosome” (Science, 2017), (iii) “RNA force field with accuracy comparable to state-of-the-art protein force fields” (PNAS, 2018; D. E. Shaw Research), (iv) “Predicting site-binding modes of ions and water to nucleic acids using molecular solvation theory” (JACS, 2019), (v) “RIC-seq for global in situ profiling of RNA-RNA spatial interactions” (Nature, 2020), and (vi) “DNA mismatches reveal conformational penalties in protein-DNA recognition” (Nature, 2020).
Broad integrations. To make DSSR as widely accessible as possible, I have initiated collaborations with the principal developers of Jmol and PyMOL. The DSSR-Jmol and DSSR-PyMOL integrations bring unparalleled search capabilities (e.g., ‘select junctions’ for all multi-branch loops) and innovative visualization styles into 3D nucleic acid structures. DSSR has also been adopted into numerous other structural bioinformatics resources, including: (i) URS, (ii) RiboSketch, (iii) RNApdbee, (iv) forgi, (v) RNAvista, (vi) VeriNA3d, (vii) RNAMake, (viii) ElTetrado, (ix) DNAproDB, (x) LocalSTAR3D, (xi) IPANEMAP, and (xii) RNANet.
Advanced features. DSSR may be licensed from Columbia University. DSSR Pro is the commercial version. It has more functionalities than DSSR basic (the free academic version), including: (i) homology modeling via in silico base mutations, a feature employed by Merck scientists, (ii) easy generation of regular helical models, including circular or super-helical DNA (see figures below), (iii) creation of customized structures with user-specified base sequences and rigid-body parameters, (iv) efficient processing of molecular dynamics (MD) trajectories, (v) detailed characterization of DNA-protein or RNA-protein spatial interactions, and (vi) template-based modeling of DNA-protein complexes (see figures below). DSSR Pro supersedes 3DNA. It integrates the disparate analysis and modeling programs of 3DNA under one umbrella, and offers new advanced features, through a convenient interface. For example, with the mutate module of DSSR Pro, one can automatically perform the following tasks: (i) mutate all bases to Us, (ii) mutate bases in hairpin loops to Gs, and (iii) mutate G–C Watson-Crick pairs to C–G, and A–U to U–A. Moreover, DSSR Pro includes an in-depth user manual and one-year technical support from the developer.
Quality control. DSSR is a solid software product that excels in RNA structural bioinformatics. It is written in strict ANSI C, as a single command-line program. It is self-contained, with zero runtime dependencies on third-party libraries. The binary executables for macOS, Linux, and Windows are just ~2MB. DSSR has been extensively tested using all nucleic-acid-containing structures in the PDB. It is also routinely checked with Valgrind to avoid memory leaks. DSSR requires no set up or configuration: it simply works.

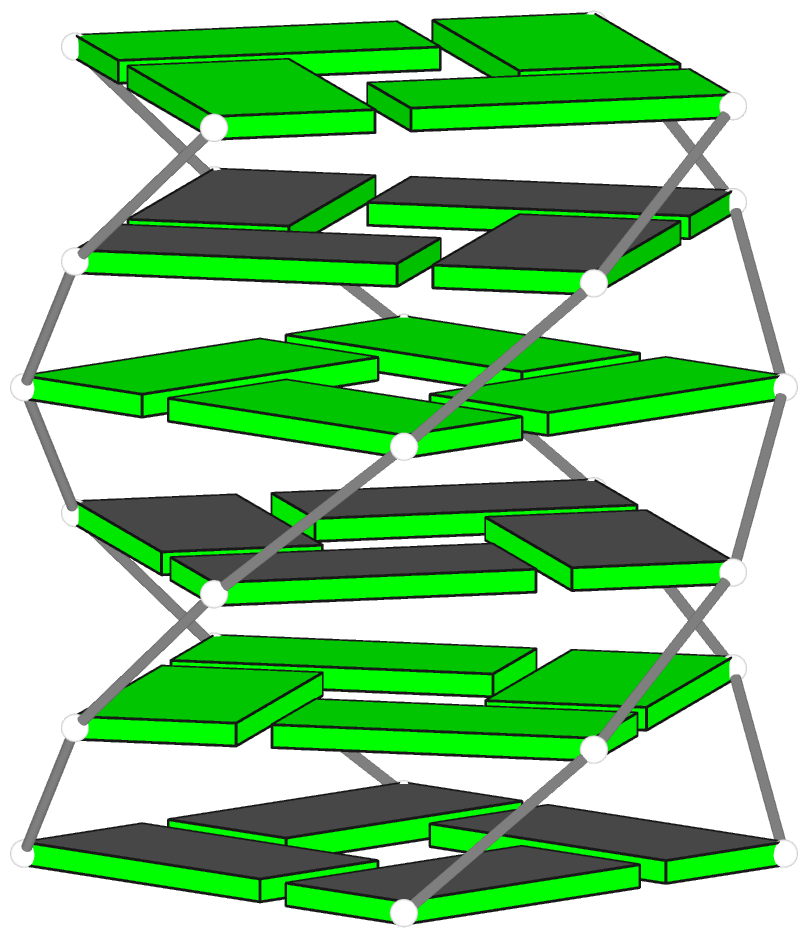
Theoretical models of G-quadruplexes, created using DSSR Pro.

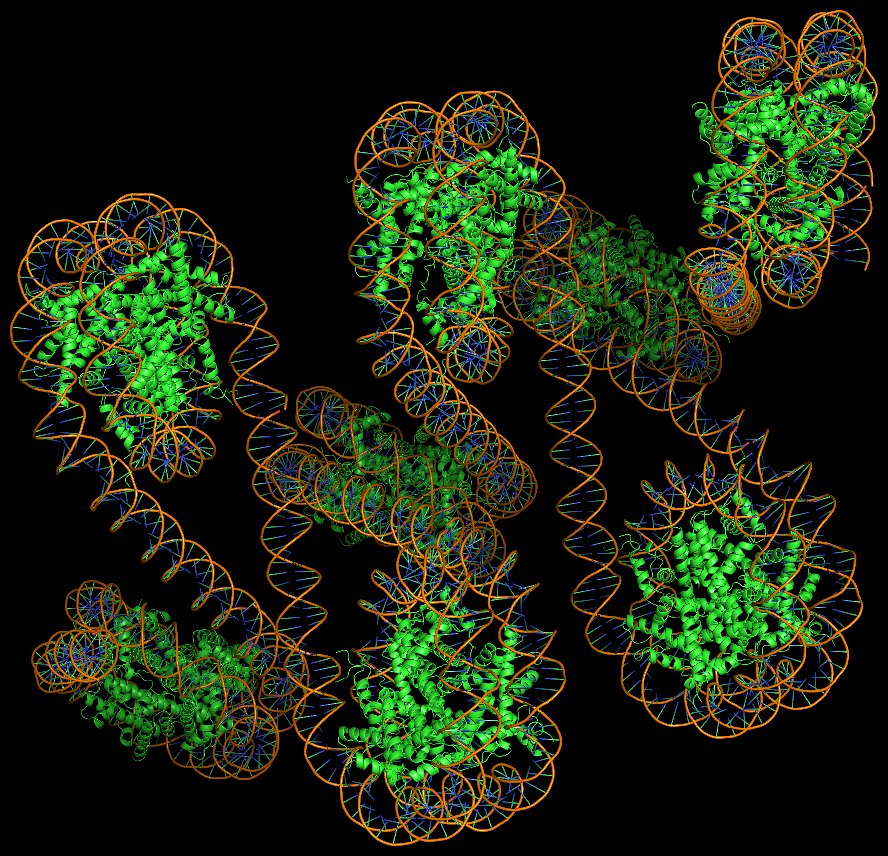
Template-based modeling of DNA-protein complexes using DSSR Pro.
Here are two chromatin-like models using PDB entry 4xzq as the template.


Circular DNA duplexes modeled using DSSR Pro.
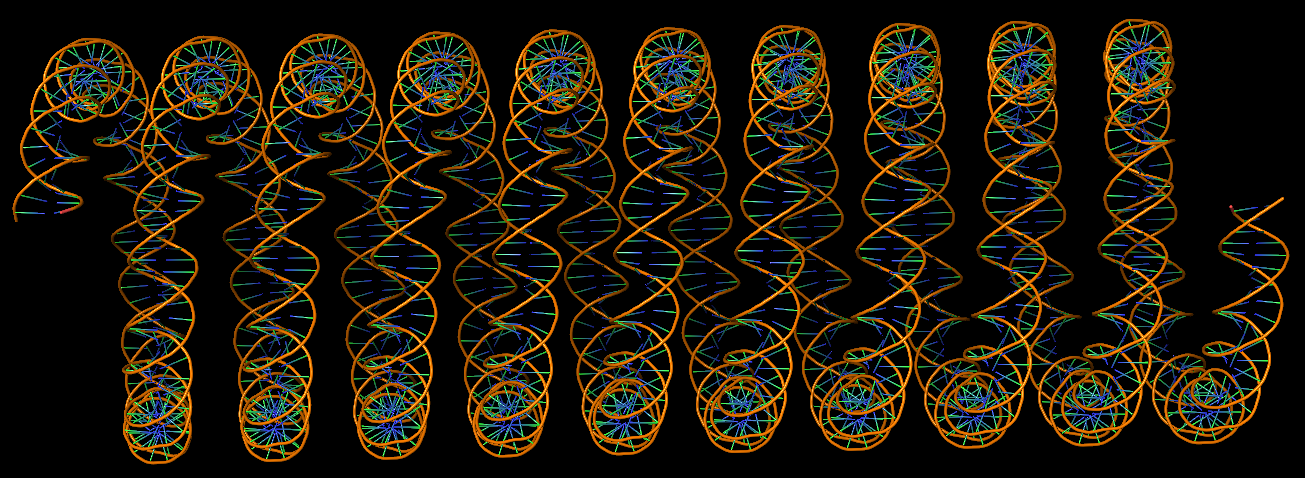
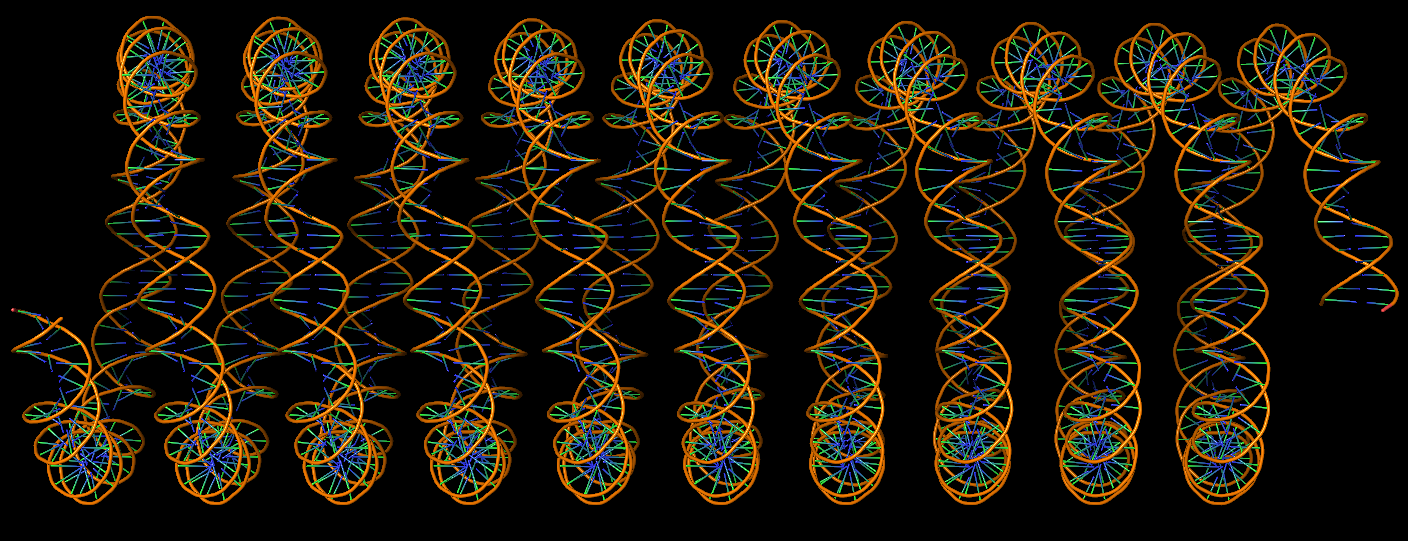
DNA super helices modeled using DSSR Pro.

Innovative cartoon-block schematics enabled by the DSSR-PyMOL integration for six representative PDB entries. Watson-Crick pairs are shown as long blocks with minor-groove edges in black (A, B), G-tetrads represented as square blocks and the metal ion as sphere ©, the ligand rendered as balls-and-sticks (D), and proteins depicted as purple cartoons (E, F). Color code for base blocks: A, red; C, yellow; G, green; T, blue; U, cyan; G-tetrad, green; WC-pairs, per base in the leading strand. Visit http://skmatic.x3dna.org.
Recommended in Faculty Opinions: “simple and effective”, “Good for Teaching”.
Employed by the NDB to create cover images of the RNA Journal.
Via PDB weekly update, I recently came across PDB entry 6neb, which is solved by NMR and described as an “MYC promoter G-quadruplex with 1:6:1 loop length”. I downloaded the atomic coordinates of the entry and ran DSSR on it. Indeed, DSSR readily identifies a three-layered parallel G-quadruplex (G4) with three propeller-type loops of 1, 6 and 1 nucleotides (i.e., 1:6:1), as shown below.
List of 1 G4-stem
Note: a G4-stem is defined as a G4-helix with backbone connectivity.
Bulges are also allowed along each of the four strands.
stem#1[#1] layers=3 INTRA-molecular loops=3 descriptor=3(-P-P-P) note=parallel(4+0) UUUU parallel
1 glyco-bond=---- groove=---- WC-->Major nts=4 GGGG A.DG3,A.DG7,A.DG16,A.DG20
pm(>>,forward) area=14.54 rise=3.36 twist=24.7
2 glyco-bond=---- groove=---- WC-->Major nts=4 GGGG A.DG4,A.DG8,A.DG17,A.DG21
pm(>>,forward) area=9.67 rise=3.48 twist=30.3
3 glyco-bond=---- groove=---- WC-->Major nts=4 GGGG A.DG5,A.DG9,A.DG18,A.DG22
strand#1 U DNA glyco-bond=--- nts=3 GGG A.DG3,A.DG4,A.DG5
strand#2 U DNA glyco-bond=--- nts=3 GGG A.DG7,A.DG8,A.DG9
strand#3 U DNA glyco-bond=--- nts=3 GGG A.DG16,A.DG17,A.DG18
strand#4 U DNA glyco-bond=--- nts=3 GGG A.DG20,A.DG21,A.DG22
loop#1 type=propeller strands=[#1,#2] nts=1 A A.DA6
loop#2 type=propeller strands=[#2,#3] nts=6 TTTTAA A.DT10,A.DT11,A.DT12,A.DT13,A.DA14,A.DA15
loop#3 type=propeller strands=[#3,#4] nts=1 T A.DT19
I then read the associated paper titled Solution Structure of a MYC Promoter G-Quadruplex with 1:6:1 Loop Length lately published in the new, open-access ACS Omega journal. The reported structure 6neb has a 27-nt sequence (termed Myc1245) of bases 5’-TTGGGGAGGGTTTTAAGGGTGGGGAAT-3’. Myc1245 is based on the 27-nt long, purine-rich MycPu27 which has 5 tracts of guanines of G4-forming motif within the MYC promoter. In Myc1245, the third G-tract of MycPu27 has been replaced by TTTA, thus it uses only G-tracts 1, 2, 4, 5 for G4 formation. Previously, it was shown that Myc2345 (using G-tracts 2-5 of MycPu27) adopts a parallel G4 structure with three propeller loops of 1:2:1 nt length.
The MycPu27 sequence is representative of the G4-forming nuclease hypersensitive element (NHE III1) within the promoter region of the MYC oncogene. Formation of G4 structures suppresses MYC transcription, thus ligand-induced G4 stabilization in the DNA level is a promising strategy for cancer therapy. The NHE III1 motif can fold into multiple G4 structures depending on factors such as protein binding. The paper on 6neb illustrates that nucleolin, a protein shown to bind MYC G4 and repress MYC transcription, preferably binds the 1:6:1 loop length conformer than the 1:2:1 conformer (the major form under physiological conditions).
The DSSR analysis of 6neb shows that the two G-tetrad steps have different overlapping areas and twist angles. The top step comprising G3 and G4 (Fig. 1A) has better stacking interactions (14.5 Å2) and smaller twist (25º) than the bottom step containing G4 and G5 (9.7 Å2 and 30º, respectively).
area=14.54 rise=3.36 twist=24.7
area=9.67 rise=3.48 twist=30.3
The analysis characterizes the T1–A15 pair as a reverse Hoogsteen pair (rHoogsteen), which is distinct from the T+A Hoogsteen pair. In DSSR, the rHoogsteen pair is of type M–N (anti-parallel), whilst the Hoogsteen pair is of type M+N (parallel). With the local base-reference frames attached (Fig. 1B), it is easy to visualize that the z-axis of T1 is pointing out of the base-pair plane, and the z-axis of A15 is pointing inwards. See also my blog post Hoogsteen and reverse Hoogsteen base pairs.
Fig. 1C shows the ATG-triad automatically identified by DSSR. As is clear in Fig. 1A, the ATG-triad stacks on the 3-layered G4 structure on the 5’ side. Moreover, with color-coded base blocks (G in green, T in blue, and A in red), the two stacks (T10–T11, and T12–T13–A14) in the 6-nt central propeller loop is immediately obvious.

Figure 1. DSSR-derived structural features in PDB entry 6neb. The images were created using DSSR and PyMOL.
In the 6neb paper, the author stated that “The central loop of 6 nt connects the outer tetrads by spanning the G-core. A single nucleotide is the minimal length of this structural motif, so the five additional residues can significantly increase the loop’s conformational flexibility.” (p.2536) It is worth noting that in PDB entry 2m53, described in G-rich VEGF aptamer with locked and unlocked nucleic acid modifications exhibits a unique G-quadruplex fold, it was observed that:
An unprecedented all parallel-stranded monomeric G-quadruplex with three G-quartet planes exhibits several unique structural features. Five consecutive guanine residues are all involved in G-quartet formation and occupy positions in adjacent DNA strands, which are bridged with a no-residue propeller-type loop.
The G4 structure is polymorphic. It seems every imaginable or even unexpected form is possible, depending on the context.

In addition to the well-known G-tetrad serving as the building block of G-quadruplexes (G4), other types of homogeneous or heterogeneous base-tetrads are also possible. In DSSR, all these base tetrads are generally termed multiplets where three or more bases associate in a co-planar fashion via H-bonding interactions.
In the context of G4 structures, U-tetrads are the most common. Fig. 1A shows an example of U-tetrads in PDB entry 4rne reported in the paper titled Structural Variations and Solvent Structure of r(UGGGGU) Quadruplexes Stabilized by Sr2+ Ions.. In the structure (Fig. 1B), two terminal U-tetrads cap the six-layered G4 structure in the middle. The four U’s in the U-tetrad are paired in parallel orientation (i.e., U+U), just as the G+G pairs in the G-tetrad of G4 structures (Fig. 1C). On the other hand, there is only one H-bond (O4…N3) in the U+U pair of the U-tetrad, in contrast to the two H-bonds in the G+G pair of the G-tetrad (Fig. 1C). In the PDB entry 4rne, DSSR also detects two octads where the middle G-tetrad is surrounded by four U’s in anti-parallel orientation (G’s filled in green vs. U’s empty, see Fig. 1C for an example).
Similarly orientated C-tetrad (C+C pair, Fig. 1D) or A-tetrad (A+A pair, Fig. 1E) are also possible. PDB entry “6a85”, associated with the paper High-resolution DNA quadruplex structure containing all the A-, G-, C-, T-tetrads., reported a high-resolution crystal structure of sequence 5'-AGAGAGATGGGTGCGTT-3' which contains all the homogeneous A-, G-, C-, T-tetrads, and the heterogeneous A:T:A:T tetrads. As of this writing (Feb. 19, 2019), the status for the PDB entry “6a85” is still “HPUB” (‘processing complete, entry on hold until publication’) even though the paper was published several months ago. Using mutate_bases in 3DNA, I generated a C-tetrad and an A-tetrad as shown in Fig. 1D and 1E. As the U-tetrad (Fig. 1A), the C- and A-tetrads also have only one H-bond in their M+N type pairs. The G-tetrad, with two H-bonds in its connecting pairs, is more stable than the other homogeneous base tetrads, leading to wide-spread G4 structures.
In the homogeneous base tetrads shown in Fig.1A-E, pairs are of the parallel M+N type and the bases are associated via their Watson-Crick and major-groove (Hoogsteen) edges. Two canonical (WC or G—U wobble) pairs can also associate via their minor-groove edges, as seen in PDB entries 2hk4 and 2lsx. Fig. 1F gives an example with two G—U wobble pairs (of anti-parallel M—N type, filled U in blue vs. empty G) in PDB entry 2lsx reported in the paper titled A minimal i-motif stabilized by minor groove G:T:G:T tetrads..

Figure 1. Non-G base tetrads automatically identified or modeled by 3DNA-DSSR. The images were created using DSSR and PyMOL.
A G-quadruplex (G4) is composed of stacks of G-tetrad where four guanines form four G•G pairs in a circular, planar fashion. Specifically, the G•G pairs of the G-tetrad (see Fig. 1A below) in G4 are of type M+N according to 3DNA/DSSR: i.e., G+G with the local z-axes of pairing guanines in parallel. Moreover, the G+G pair is uniquely quantified by three base-pair parameters: Shear, Stretch, and Opening with mean values [+1.6 Å, +3.5 Å, –90º] or [–1.6 Å, –3.5 Å, +90º], corresponding to the cWH (cW+M) or cHW (cM+W) types of LW (DSSR) classifications, respectively. This pair is numbered VI in the list of 28 base pairs with two or more H-bonds between base atoms, compiled by Saenger.
In addition to the standard G-tetrad configuration as normally seen in G4 structures, a so-called pseudo-G-tetrad form (see Fig. 1B below) is reported in a 2013 paper titled Duplex-quadruplex motifs in a peculiar structural organization cooperatively contribute to thrombin binding of a DNA aptamer. (PDB entry 4i7y). In a 2017 publication from the same group, Through-bond effects in the ternary complexes of thrombin sandwiched by two DNA aptamers, another form of pseudo-G-tetrad (Fig. 1C) is found in PDB entries 5ew1 and 5ew2.
Clearly, pseudo-G-tetrads are very different from the normal G-tetrad, in terms of base pairing patterns. The G-tetrad is highly regular with the same type of G+G pairs, with the O6 atoms pointing to the middle of the circle. The two pseudo-G-tetrads are less regular, and they differ from each other as well, by flipping G12 from syn (Fig. 1B) to trans (Fig. 1C).
These distinctions stand out even more by filling the up-face (+z-axis outwards) of a guanine base in green while leaving the down-face (+z-axis inwards) empty (G5 in Fig. 1B, G5 and G12 in Fig. 1C). So in G-tetrad (Fig. 1A), all four guanines have their positive z-axis point towards the viewer, corresponding to all four G+G pairs. In one pseudo-G-tetrad (Fig. 1B), G5 has its positive z-axis pointing away from the viewer. So G5–G7 and G5–G16 pairs are of the M–N type. The other type of pseudo-G-tetrad (Fig. 1C) has the opposite orientation for G12. Finally, Fig. 1D shows schematically PDB entry 4i7y where the G-tetrad and a pseudo-G-tetrad are directly stacked, creating a two-layered pseudo-G-quadruplex.

Figure 1. (A) G-tetrad, (B-C) two types of pseudo-G-tetrads, and (D) the complex of a DNA-apatmer with thrombin. G-tetrads were automatically identified by 3DNA-DSSR. The images were created using DSSR and PyMOL.
A starting structure of suitable sequence is a prerequisite for many applications, including downstream use in X-ray crystallography, NMR, and molecular dynamics (MD) simulations. Browsing through the literature, I’ve noticed the following tools for such a purpose.
- The make-na server, a web-based automated tool for making nucleic acid helices powered by NAB. It supports abasic sites via the underscore character (
_). According to the help page, “The structure file represents the abasic as the 3-letter code ‘3DR’ in DNA strands and ‘ N’ in RNA strands. These are Protein Data Bank conventions.” An example input is shown below:
ATACCGATACG_TAGAC
TG_CTATGCTATCTGT_
- The NAB itself, and the standalone fd_helix.c program which supports 6 fiber-based models of DNA or RNA.
- The NUCGEN program from the Bansal group. “The NUCGEN software generates double helical models with the backbone fixed in B-form DNA, but with appropriate modifications in the input data, it can also generate A-form DNA and RNA duplex structures.”
- 3DNA and its web interface. The ‘rebuild’ program can be used for constructing customized, single or duplex DNA/RNA structures based on a set of base-pair and step (helical) parameters. Moreover, the sugar-phosphate backbone in A-, B- or RNA conformation is allowed. The ‘fiber’ program incorporates a comprehensive list of 56 regular models, based mostly on fiber diffraction data. The list includes single, duplex, triplex, quadruplex, DNA, RNA structures or their hybrids. Notable, the classic Pauling’s triplex model is also available. The 3DNA web 2.0 makes these model-building features readily accessible to a large user base.
Overall, each of the tools listed above has its unique features and may fit better for different applications. It is to the benefit of the user community to have a choice.
Nowadays, I’ve been used to google searches as a quick way to solve problems. Once in a while, I come across a tip or trick that fixes an issue at hand and then move on. However, I may late on meet a similar problem, but only vaguely remember how I solved it previously. So I’d need to start googling around again. This list is a remedy for such situations, and it will be continuously updated. While the list is created for my own reference, it may also be useful to other viewers of the post, presumably reaching here via google.
icdiff — show diff with colorscc — strip C commentsTaskwarrior (taks) — manage TODO list from terminalhttpie as a replacement of curl and wgetbyebug and pry for debugging Rubyag to search for PATTERN in source files, replacing grepfd to find files and directoriesbat to view files with syntax highlighting (in place of cat)exa as an alternative to lsbench to benchmark codeasciinema and svg-term to record terminal activity as an SVG animation. Another option is termtosvg. Moreover, the trio ttyrec, ttyplay, ttygif can record, play terminal screen recordings, and convert it into smooth GIFwrk to benchmark HTTP APIshub — git wrapper for GitHubtail -n +2 to skip the first line (starting from the second line)sudo -i -u user_id, the -i or --login option invokes login shell- Understanding Shell Script’s idiom: 2>&1 — redirect ‘stderr’ to ‘stdout’ via ’2>&1’ in
bash shell.
- Ruby one-liners
ruby -pi.bak -e "gsub(/SOME_PATTERN/, 'other_text')" files for global replacement of SOME_PATTERN by other_text in filesruby -pe 'gsub(/_/, ".")' globally replace ‘_’ with ‘.’
Recently, a 3DNA user asked on the Forum about how to perform mutations to 3-methyladenine. The user reported that the procedure described in the FAQ entry How can I mutate cytosine to 5-methylcytosine did not work for the case of 3-methyladenine. This ‘limitation’ is easily understandable: the 3DNA mutate_bases program must have knowledge of the target base, 3-methyladenine, to perform the mutation properly. The program works for the most common 5-methylcytosine mutations since the corresponding 5MC file (Atomic_5MC.pdb, in the standard base-reference frame) is already included within the 3DNA distribution. By supplying a similar file for the target base, mutate_bases runs the same for mutations to 5-methylcytosine (or other bases). This blog post outlines the procedure, using 3-methyladenine as an example.
A ligand name search for 5-methylcytosine on the RCSB PDB led to only two matched entries: 2X6F and 3MAG. The ligand id is 3MA. Since 3MAG has a better resolution (1.8 Å) than 2X6F (3.3 Å), its 3MA ligand was extracted from the corresponding PDB file (3MAG.pdb). The atomic coordinates, excluding those for the two hydrogens, are as below. Note that the 3-methyl carbon atom is named CN3.
HETATM 2960 N9 3MA A 600 16.587 14.258 22.170 1.00 49.87 N
HETATM 2961 C4 3MA A 600 17.123 13.100 21.622 1.00 50.46 C
HETATM 2962 N3 3MA A 600 16.877 11.811 22.009 1.00 50.37 N
HETATM 2963 CN3 3MA A 600 15.983 11.363 23.063 1.00 50.41 C
HETATM 2964 C2 3MA A 600 17.590 10.968 21.241 1.00 50.11 C
HETATM 2965 N1 3MA A 600 18.422 11.217 20.224 1.00 49.27 N
HETATM 2966 C6 3MA A 600 18.627 12.484 19.858 1.00 48.99 C
HETATM 2967 N6 3MA A 600 19.426 12.709 18.829 1.00 46.12 N
HETATM 2968 C5 3MA A 600 17.949 13.503 20.593 1.00 49.89 C
HETATM 2969 N7 3MA A 600 17.929 14.900 20.488 1.00 49.84 N
HETATM 2970 C8 3MA A 600 17.113 15.286 21.434 1.00 49.58 C
After running the 3DNA utility program std_base with options -fit -A, the corresponding atomic coordinates of 3MA are transformed to the standard base reference frame of adenine. The file must be named Atomic_3MA.pdb, and it has the following contents:
HETATM 1 N9 3MA A 1 -1.287 4.521 0.006 1.00 49.87 N
HETATM 2 C4 3MA A 1 -1.262 3.133 0.004 1.00 50.46 C
HETATM 3 N3 3MA A 1 -2.337 2.286 -0.009 1.00 50.37 N
HETATM 4 CN3 3MA A 1 -3.743 2.648 -0.047 1.00 50.41 C
HETATM 5 C2 3MA A 1 -1.905 1.013 0.001 1.00 50.11 C
HETATM 6 N1 3MA A 1 -0.662 0.520 0.004 1.00 49.27 N
HETATM 7 C6 3MA A 1 0.366 1.372 -0.003 1.00 48.99 C
HETATM 8 N6 3MA A 1 1.588 0.867 -0.034 1.00 46.12 N
HETATM 9 C5 3MA A 1 0.068 2.768 0.003 1.00 49.89 C
HETATM 10 N7 3MA A 1 0.875 3.914 -0.003 1.00 49.84 N
HETATM 11 C8 3MA A 1 0.026 4.909 -0.003 1.00 49.58 C
Note that in file Atomic_3MA.pdb, (1) the z-coordinates of the base atoms are close to zeros, (2) the ordering of atoms is as in the original ligand of 3MA shown above.
With Atomic_3MA.pdb in place (in the current working directory, or the $X3DNA/config folder), one can perform 3-methyladenine mutations using mutate_bases. For illustration purpose, let’s generate a B-form DNA with base sequence GACATGATTGCC using the 3DNA fiber program:
fiber -seq=GACATGATTGCC fiber-BDNA.pdb
To mutate A7 to 3MA, one needs to run mutate_bases as following:
mutate_bases "chain=A s=7 m=3MA" fiber-BDNA.pdb fiber-BDNA-A7to3MA.pdb
The result of the mutation is shown in the figure below. Note that the backbone has identical geometry as that before the mutation, and the mutated 3MA-T pair has exactly the same parameters (propeller/buckle etc) as the original A-T. These are the two defining features of the 3DNA mutate_bases program.

Please see the thread mutations to 3-methyladenine on the 3DNA Forum to download files fiber-BDNA.pdb and fiber-BDNA-A7to3MA.pdb.
When visiting the RCSB PDB website today, I am please to notice that the PDB now contains “10015 Nucleic Acid Containing Structures”. Based on “Macromolecule Type” in “Advanced Search” of the RCSB PDB website, I observed the following information:
- The number of DNA-containing structures is
6,384 (reported in 2,997 papers), and the corresponding number for RNA-containing structures is 3,861 (associated with 2,012 publications).
- There are
4,570 structures containing both DNA and protein (potentially forming DNA-protein complexes), and 2,478 RNA-protein complexes.
- The smallest nucleic-acid-containg structures have only two nucleotides (e.g., 3rec), and largest ones are the ribosomes (and virus particles).
- The earliest released DNA structure from the PDB is 1zna (on March 18, 1981), a Z-DNA tetramer. The earliest RNA structure released is 4tna (on April 12, 1978), a refined structure of the yeast phenylalanine transfer RNA.
This landmark achievement is made possible by the world-wide scientific community through decades of efforts solving DNA/RNA 3D structures via experimental approaches (mainly solution NMR, x-ray crystal, and cryo-EM). These over 10K nucleic acid structures present both challenges and opportunities for the field of structural bioinformatics, especially for intricate RNA molecules. DSSR is an integrated software tool for dissecting the spatial structure of RNA. It is my effort in addressing the challenging issues for the analysis/annotation and visualization of RNA structures.
It is textbook knowledge that the Watson-Crick (WC) pairs are specific, forming only between A and T/U (A–T/U or T/U–A) or G and C (G–C or C–G). Furthermore, an A only forms one WC pair with a T, so is G vs. C. The widely used dot-bracket-notation (DBN) of DNA/RNA secondary structure depends crucially on this feature of specificity and uniqueness, by using matched parentheses to represent WC pairs, such as ((....)) for a GCGA (GNRA-type) tetra-loop of sequence GCGCGAGC.
The reality is more complicated, even for what’s presumably to be a ‘simple’ question of deriving RNA secondary structure from 3D coordinates in PDB. One subtlety is related to the ambiguity of atomic coordinates that renders one base apparently forming two WC pairs with two other complementary bases. As always, the case can be best illustrated with a concrete example. The image shown below is taken from PDB entry 1qp5 where C20 (on chain B) forms two WC pairs, each with G4 and G5 (on chain A) respectively.

Clearly, taking both as valid WC G–C pairs would make the resultant DBN illegitimate. DSSR resolves such discrepancies by taking structural context into consideration to ensure that one base can only have a WC pair with another base. Here the G5–C20 WC pair is retained whilst the G4–C20 WC is removed.
This issue, one base can form two WC pairs as derived from the PDB, has been noticed for a long while. Two examples from literature are shown below:
The crystal structure data files were downloaded from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (Berman et al. 2000). For each crystal structure, the set of canonical base pairs was extracted by selecting all Watson–Crick and standard G-U wobble pairs found by RNAview (Yang et al. 2003). Occasional conflicts in this list, where RNAview annotates two bases, x and y, as a standard base pair and also y and z as another conflicting base pair, were removed manually by visual inspection of the crystal structure in the program PyMOL (http://pymol.sourceforge.net/). The helix-extension data set was created by taking the canonical pairs and adding all additional base–base interactions identified by RNAview (excluding stacked bases and tertiary interactions) for which the direct neighbor was already in the collection. This means each base pair (i,j) was added if both i and j were still unpaired and if either (i + 1, j – 1) or (i –1, j + 1) were already in the set.
… From these complexes, we retrieved all RNA chains also marked as non-redundant by RNA3DHub. Each chain was annotated by FR3D. Because FR3D cannot analyze modified nucleotides or those with missing atoms, our present method does not include them either. If several models exist for a same chain, the first one only was considered. For the rest of this paper, the base pairs extracted from the FR3D annotations are those defined in the Leontis–Westhof geometric classification (24).
For each chain a secondary structure without pseudoknots was deduced from the annotated interactions, as follows. First all canonical Watson–Crick and wobble base pairs (i.e. A-U, G-C and G-U) were identified. Then, since many structures are naturally pseudoknotted, we used the K2N (25) implementation in the PyCogent (26) Python module to remove pseudoknots. Problems arise when a nucleotide is involved in several Watson–Crick base pairs (which is geometrically not feasible), probably due to an error of the automatic annotation. Those discrepancies were removed with a ad hoc algorithm such that if a nucleotide is involved in several Watson–Crick base pairs, we remove the base pair which belongs to the shortest helix.
By design, DSSR takes care of these ‘little details’, among other handy features (such as handling modified nucleotides and removing pseudoknots). By providing a robust infrastructure and comprehensive framework, DSSR allows users to focus on their research topics. If you have experience with other tools, such as RNAView and FR3D cited above, give DSSR a try: it may fit your needs better.
An article titled Simulations and electrostatic analysis suggest an active role for DNA conformational changes during genome packaging by bacteriophages has recently been published in bioRxiv. I was honored to have the opportunity collaborating with fellow researchers from University of Pennsylvania and Thomas Jefferson University in this significant piece of work.
Here is the abstract. Please download the PDF version to know more.
Motors that move DNA, or that move along DNA, play essential roles in DNA replication, transcription, recombination, and chromosome segregation. The mechanisms by which these DNA translocases operate remain largely unknown. Some double-stranded DNA (dsDNA) viruses use an ATP-dependent motor to drive DNA into preformed capsids. These include several human pathogens, as well as dsDNA bacteriophages (viruses that infect bacteria). We previously proposed that DNA is not a passive substrate of bacteriophage packaging motors but is, instead, an active component of the machinery. Computational studies on dsDNA in the channel of viral portal proteins reported here reveal DNA conformational changes consistent with that hypothesis. dsDNA becomes longer (“stretched”) in regions of high negative electrostatic potential, and shorter (“scrunched”) in regions of high positive potential. These results suggest a mechanism that couples the energy released by ATP hydrolysis to DNA translocation: The chemical cycle of ATP binding, hydrolysis and product release drives a cycle of protein conformational changes. This produces changes in the electrostatic potential in the channel through the portal, and these drive cyclic changes in the length of dsDNA. The DNA motions are captured by a coordinated protein-DNA grip-and-release cycle to produce DNA translocation. In short, the ATPase, portal and dsDNA work synergistically to promote genome packaging.
{kind=link}