Since securing funding for the X3DNA-DSSR project through an NIH R24 grant, I have dedicated myself to continuously advancing and refining the software tool. DSSR is a robust and mature tool with a professional user manual. Getting it up and running is straightforward, and assistance for installation is rarely required. Common usages are also already addressed, allowing me to focus on developing new features and addressing edge cases proactively.
- I regularly test DSSR using the latest weekly updates from the Protein Data Bank (PDB), identifying and resolving issues before users report them.
- I actively monitor the 3DNA Forum, providing timely responses to user queries, addressing reported issues, and introducing new features when necessary.
- Writing papers and blog posts can be effective methods to highlight areas where clarification and improvement are needed.
- Collaborating with other researchers often leads to enhancements in DSSR.
- I monitor how DSSR is cited, respond quickly to any reported issues, and contact authors when necessary.
As an example, I noticed a recent article titled 'Structural Analysis of Uridine Modifications in Solved RNA Structures' (https://doi.org/10.1093/nargab/lqaf197) by Arteaga and Znosko, where DSSR was cited as below:
Secondary structure elements (SSEs) containing uridine modifications were identified and annotated using Dissecting the Spatial Structure of RNA (DSSR). Corresponding SSEs containing canonical uridine were identified via RNA Characterization of Secondary Structure Motifs (CoSSMos) and also annotated using DSSR.
Identification and characterization of SSEs RNA SSEs were identified and characterized using the software Disecting the Spatial Structure of RNA (DSSR) [76]. DSSR, an RNA-specific successor to 3DNA [77], was employed to analyze RNA structures containing U modifications. The analysis was performed using default parameters, and all output data were saved in a ∗.json file format. For each ∗.json file, relevant information regarding the U modification residues was extracted. This information included the type of SSE, its associated nucleotide sequence, hydrogen bond acceptor/donor groups, base stacking (π stacking) interactions, sugar pucker, and glycosidic angle. The distance cutoff for identifying hydrogen bonding and base stacking interactions was set to 4.0 Å, as per DSSR’s default settings.
The following citation draw my attention:
An additional 21 structures were excluded from the analysis due to missing atoms in the RCSB PDB entries, DSSR processing issues, incomplete SSEs, or the inability to clip the SSEs (Supplementary Table S2).
I am curious to understand the DSSR processing issues referred to in their study. Upon reviewing 'Supplemental Table S2. Structures excluded from the analysis,' I noted that the following 15 PDB entries were listed as 'Unable to be annotated by DSSR': 4U3M, 4U3U, 4U4Q, 4U4R, 4U4U, 4U52, 4V88, 4V9O, 4V9P, 5FL8, 5TBW, 6I7V, 6SV4, 6Z1P, and 7QVP.
These are all large RNA structures, with 13 of them containing more than 10,000 nucleotides each. I am unsure which version of DSSR was used in their study; however, when using the current version (v2.7.2-2026jan12), I can process these PDB entries without encountering any issues. In cases such as these, or any issues related to 3DNA/DSSR, users are encouraged to reach out. I always aim to address inquiries promptly.
References
- Arteaga SJ, Znosko BM. Structural analysis of uridine modifications in solved RNA structures. NAR Genomics and Bioinformatics. 2026;8:lqaf197. https://doi.org/10.1093/nargab/lqaf197.

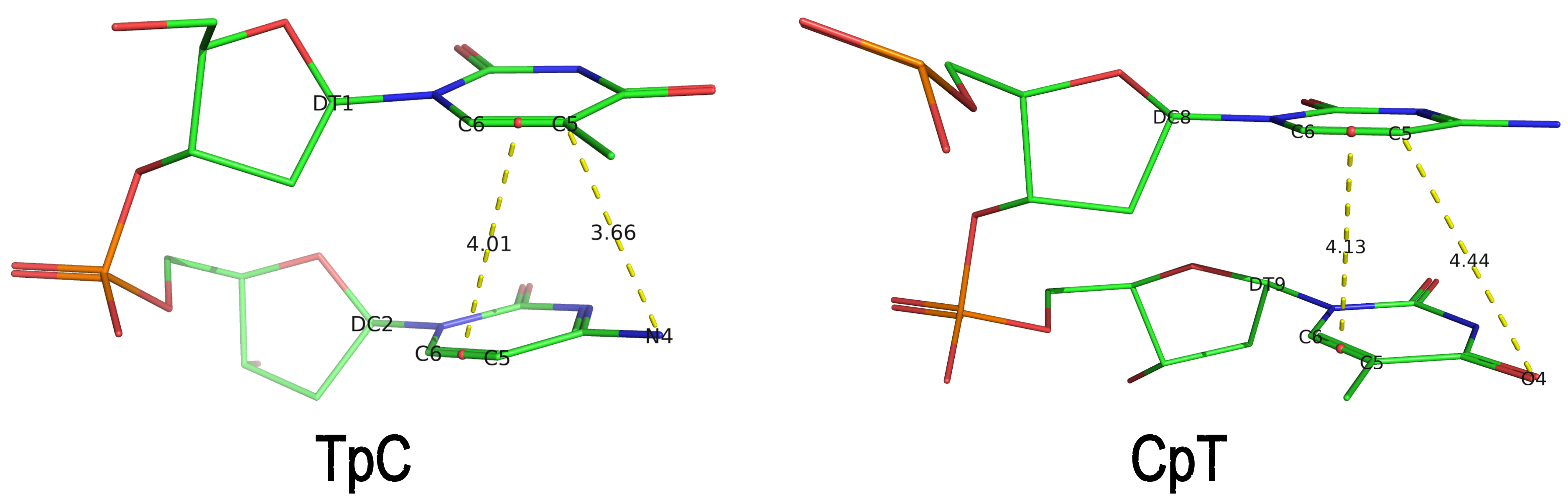
Recently, I read the preprint of Gordan et al. (2025), titled "High-throughput characterization of transcription factors that modulate UVdamage formation and repair at single-nucleotide resolution". In the METHODS section on "Structural analysis of AlphaFold 3 predicted TF-DNA complexes", the authors introduced two geometric parameters to characterize dipyrimidines, as detailed below:
Base-step d22 distance, d64 distance, of dipyrimidines were computed for each base-step per DNA strand using custom PyMOL python scripts74. d22 was defined as the distance in Ångstroms (Å) between the C5-C6 bond midpoints between adjacent pyrimidines. d64 was defined as the Å distance between the 5' pyrimidine's C5 and X4 (either O or N) attached to the 3' pyrimidine's C4.
These d22 and d64 parameters are well-defined and straightforward to calculate (see the figure below for illustrative examples). They can be integrated seamlessly with DSSR's infrastructure, requiring minimal additional coding effort. As a result, I have decided to implement them into DSSR.
For example, in the case of the MyoD bHLH domain-DNA complex (PDB ID: 1mdy), running the following DSSR (v2.7.1) command:
x3dna-dssr -i=1mdy.pdb --json -o=1mdy.json
generates a JSON file (1mdy.json), which contains the following information under the nts section for E.DT1 (connected with E.DC2): "d22": 4.014 and "d64": 3.655."
 The default human-readable output file,
The default human-readable output file, dssr-torsions.txt, now includes two additional columns for d22 and d64 under the section titled 'Main chain conformational parameters,' as shown below.
nt d22 d64
1 T E.DT1 4.01 3.66
2 C E.DC2 --- ---
3 A E.DA3 --- ---
4 A E.DA4 --- ---
5 C E.DC5 --- ---
6 A E.DA6 --- ---
7 G E.DG7 --- ---
8 C E.DC8 4.13 4.44
9 T E.DT9 --- ---
Note that pseudouridine (Ψ) is excluded from the calculation of d22 and d64 parameters of a dipyrimidine base step.
The implementation of d22 and d64 parameters in DSSR is a clear example of my proactive approach to enhancing the software's functionality. Users are always encouraged to reach out with requests for new features or improvements, as well as to report any bugs or ask questions.
References
- Gordan R, Wasserman H, Chi B, Bohm K, Duan M, Sahay H, et al. High-throughput characterization of transcription factors that modulate UVdamage formation and repair at single-nucleotide resolution. 2025. https://doi.org/10.21203/rs.3.rs-8197218/v1.
As of v2.5.4-2025jun06, DSSR automatically checks for steric clashes or exact duplicates of residues in an input coordinate file. It reports such issues instead of crashing, and will terminate only if an excessive number of overlaps are detected. An simplified example is shown below, which contains two nucleotides (G#1) on chains 0 and 1, respectively
ATOM 1 OP3 G 0 1 -4.270 51.892 37.186 1.00 27.93 O
ATOM 2 P G 0 1 -3.834 50.887 37.436 1.00 28.61 P
ATOM 3 OP1 G 0 1 -4.601 49.700 37.549 1.00 27.02 O
ATOM 4 OP2 G 0 1 -4.061 52.011 36.684 1.00 25.80 O
ATOM 5 O5' G 0 1 -2.906 51.105 38.691 1.00 28.01 O
ATOM 6 C5' G 0 1 -1.941 52.126 38.781 1.00 26.76 C
ATOM 7 C4' G 0 1 -1.037 51.914 39.967 1.00 26.12 C
ATOM 8 O4' G 0 1 -1.822 51.894 41.184 1.00 24.21 O
ATOM 9 C3' G 0 1 -0.285 50.591 39.988 1.00 25.12 C
ATOM 10 O3' G 0 1 0.884 50.614 39.172 1.00 26.09 O
ATOM 11 C2' G 0 1 0.008 50.411 41.462 1.00 26.05 C
ATOM 12 O2' G 0 1 1.102 51.209 41.880 1.00 27.46 O
ATOM 13 C1' G 0 1 -1.271 50.952 42.083 1.00 28.40 C
ATOM 14 N9 G 0 1 -2.272 49.904 42.329 1.00 27.27 N
ATOM 15 C8 G 0 1 -3.470 49.733 41.686 1.00 26.55 C
ATOM 16 N7 G 0 1 -4.137 48.712 42.125 1.00 25.36 N
ATOM 17 C5 G 0 1 -3.332 48.176 43.118 1.00 25.64 C
ATOM 18 C6 G 0 1 -3.529 47.056 43.955 1.00 24.98 C
ATOM 19 O6 G 0 1 -4.492 46.284 43.991 1.00 24.56 O
ATOM 20 N1 G 0 1 -2.460 46.862 44.821 1.00 24.78 N
ATOM 21 C2 G 0 1 -1.346 47.639 44.878 1.00 24.96 C
ATOM 22 N2 G 0 1 -0.417 47.298 45.782 1.00 23.72 N
ATOM 23 N3 G 0 1 -1.145 48.689 44.109 1.00 25.74 N
ATOM 24 C4 G 0 1 -2.171 48.901 43.257 1.00 26.32 C
ATOM 1 OP3 G 1 1 -6.437 51.060 40.254 1.00 27.81 O
ATOM 2 P G 1 1 -5.327 50.209 39.884 1.00 28.55 P
ATOM 3 OP1 G 1 1 -5.668 48.792 39.652 1.00 26.90 O
ATOM 4 OP2 G 1 1 -4.838 51.036 38.808 1.00 25.57 O
ATOM 5 O5' G 1 1 -4.301 50.297 41.090 1.00 27.94 O
ATOM 6 C5' G 1 1 -3.427 51.393 41.257 1.00 26.67 C
ATOM 7 C4' G 1 1 -2.528 51.168 42.443 1.00 26.12 C
ATOM 8 O4' G 1 1 -3.335 50.964 43.624 1.00 24.16 O
ATOM 9 C3' G 1 1 -1.648 49.928 42.372 1.00 25.13 C
ATOM 10 O3' G 1 1 -0.467 50.136 41.599 1.00 26.15 O
ATOM 11 C2' G 1 1 -1.372 49.649 43.835 1.00 25.96 C
ATOM 12 O2' G 1 1 -0.375 50.515 44.354 1.00 27.37 O
ATOM 13 C1' G 1 1 -2.714 50.006 44.458 1.00 28.21 C
ATOM 14 N9 G 1 1 -3.608 48.845 44.581 1.00 27.06 N
ATOM 15 C8 G 1 1 -4.771 48.614 43.895 1.00 26.37 C
ATOM 16 N7 G 1 1 -5.340 47.496 44.226 1.00 25.18 N
ATOM 17 C5 G 1 1 -4.502 46.957 45.190 1.00 25.44 C
ATOM 18 C6 G 1 1 -4.599 45.755 45.923 1.00 24.77 C
ATOM 19 O6 G 1 1 -5.480 44.892 45.864 1.00 24.39 O
ATOM 20 N1 G 1 1 -3.532 45.594 46.796 1.00 24.63 N
ATOM 21 C2 G 1 1 -2.504 46.469 46.949 1.00 24.81 C
ATOM 22 N2 G 1 1 -1.560 46.145 47.845 1.00 23.58 N
ATOM 23 N3 G 1 1 -2.396 47.594 46.280 1.00 25.56 N
ATOM 24 C4 G 1 1 -3.422 47.779 45.423 1.00 26.12 C
Running DSSR on the above coordinates will show the following output:
[i] 0.G1 and 1.G1 in clashes: min_dist=0.57
where min_dist refers to the minimum distance between heavy atoms of the two nucleotides.
The clash-detection feature in DSSR was added in response to the bioRxiv preprint by Kretsch et al. (2025), titled "Assessment of nucleic acid structure prediction in CASP16" (https://doi.org/10.1101/2025.05.06.652459), which noted that in some predicted RNA models submitted to CASP16, multiple models were not properly delineated with MODEL/ENDMDL in PDB format or _atom_site.pdbx_PDB_model_num in mmCIF format. I communicated with the authors, who kindly provided the PDB files to help debug the issue. For more details, see the blog post Improving DSSR through extreme cases from early June 2025 at https://x3dna.org/highlights/improving-dssr-through-extreme-cases.
The bioRxiv paper by Kretsch et al. was recently published in Proteins: Structure, Function, and Bioinformatics. The relevant citation to DSSR is in Section 2.8 | Secondary Structure Analysis, as follows:
Secondary structures were extracted from CASP16 models with DSSR (v1.9.9-2020feb06) [47]. Some models, in particular due to large clashes, could not be processed by DSSR (Table S1). The base-pair list was extracted from the table in the output file directly because the dot-bracket structure produced by DSSR, in particular for multimers, contained errors. The canonical base pairs were defined as those labeled as Watson-Crick-Franklin (WC) and wobble base pairs (hereafter referred to as ‘base pairs’ or ‘pairs’). All other base pairs are defined as non-canonical base pairs and analyzed separately. Crossed base pairs (pseudoknots) were defined as non-nested canonical base pairs, that is, any canonical base pair (i,j) for which another canonical base pair (k,l) existed with i < k < j < l or k < i < l < j. Singlet base pairs were defined as any canonical base pair that was not part of a stem, that is, (i,j) such that there was no neighboring canonical base pair between i + 1 and j − 1 or between i − 1 and j + 1. Intermolecular base pairs were identified as any canonical base pair between nucleotides in different chains.
It is worth noting that DSSR is actively supported, and I always strive to respond to users’ questions via email or (preferably) on the 3DNA Forum quickly and concretely. If you have any questions about DSSR or need clarifications, please feel free to contact me. Additionally, I monitor 3DNA/DSSR citations in the literature and proactively address issues that come to my attention when necessary.
I recently came across the paper by Zurkowski et al. (2025), titled "Detecting polynucleotide motifs: Pentads, hexads, and beyond.". The authors introduce LinkTetrado, a software tool that is described as "the first fully automated method for detecting polyadic motifs in the three-dimensional structures of nucleic acids." I am somewhat surprised by this claim, as I believe it overlooks the 2015 DSSR paper, which includes a dedicated section on "Higher-order coplanar base associations (multiplets)" as shown below:
DSSR defines multiplets as three or more bases associated in a coplanar geometry via a network of hydrogen-bonding interactions. Multiplets are identified through inter-connected base pairs, filtered by pair-wise stacking interactions and vertical separations to ensure overall coplanarity (Supplementary Figures S1, S3, S4 and S7). The abundant A-minor motifs (33) (types I and II, Supplementary Figures S3, S4 and S7) are base triplets, the smallest multiplet. The G-tetrad motif, where four guanines are associated via four pairs in a square planar geometry, is another special case of a multiplet.
In fact, DSSR multiplets are all-encompassing, including pentads, hexads, heptads, octads, etc.
The DSSR User Manual has extensive discussions (see Section 3.2.4 "Multiplets (higher-order coplanar base associations)") and several examples of multiplets, including:
- Figure 8: The GUA triplet auto-identified by DSSR in PDB entry 1msy.
- Figure 12: Base pentad (AUAAG) auto-identified by DSSR in PDB entry 1jj2. The five nts (A306,U325,A331,A340,G345) are all within the 23S rRNA.
DSSR can successfully identify the multiplets reported in the Zurkowski et al. paper, although there may be minor differences due to variations in cutoffs and definitions. For instance, using PDB ID 6w9p (shown in Fig. 7F of the Zurkowski et al. paper), DSSR can perform the following:
x3dna-dssr -i=6w9p.pdb -o=6w9p.out
x3dna-dssr -i=dssr-multiplets.pdb --select-model=4 -o=G4T3.pdb
The relevant portions of DSSR output (6w9p.out) are shown below:
List of 4 multiplets
1 nts=4 GGGG A.DG4,A.DG10,A.DG16,A.DG22
2 nts=4 GGGG A.DG5,A.DG11,A.DG17,A.DG23
3 nts=4 GGGG A.DG7,A.DG13,A.DG19,A.DG25
4 nts=7 GTGTGTG A.DG6,A.DT9,A.DG12,A.DT15,A.DG18,A.DT21,A.DG24
...
2 dssr-multiplets.pdb -- an ensemble of multiplets
DSSR can further render the extracted G4T3.pdb into the following image using PyMOL:

DSSR has far more to offer than meets the eye. See the DSSR User Manual and the practical guide to DSSR-PyMOL integration for more details.
References
Lu,X.-J. et al. (2015) DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res, gkv716.
Zurkowski,M. et al. (2025) Detecting polynucleotide motifs: Pentads, hexads, and beyond. PLoS Comput Biol, 21, e1013633.
Recently, I noticed that a user had uploaded a file to the website "DSSR-enabled Innovative Schematics of 3D Nucleic Acid Structures with PyMOL", which DSSR reported as 'no nucleotides found.' Upon visualizing it in PyMOL, the structure appeared to be a single-stranded RNA. Further investigation revealed that while the uploaded file was in PDB format, it did not adhere to the standard naming conventions for nucleotides typically used in RCSB PDB entries. For instance, an A nucleotide extracted from the file had its exocyclic amino group named as N553 instead of the conventional N6 (see below).
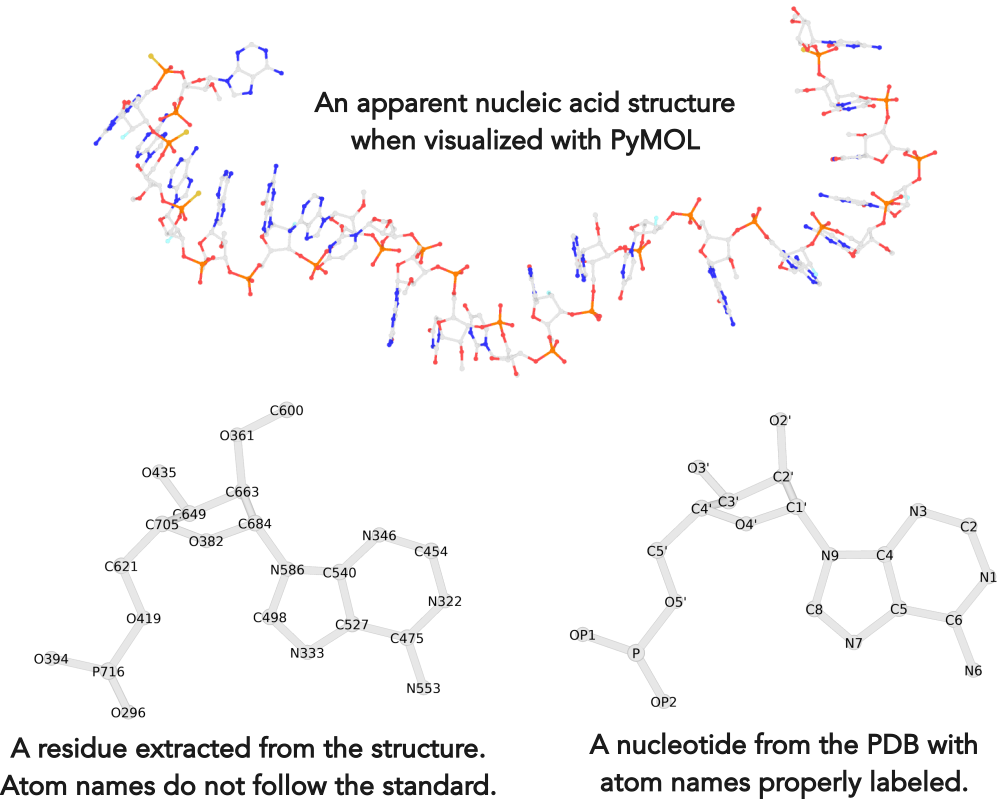
Following 3DNA, DSSR uses the atomic coordinates and standard names of base-ring atoms to identify a nucleotide. All known nucleotides share a common six-membered pyrimidine ring, with atoms named consecutively (N1, C2, N3, C4, C5, C6), and purines include three additional atoms (N7, C8, N9). See below for the standard names in Watson-Crick base pairs.
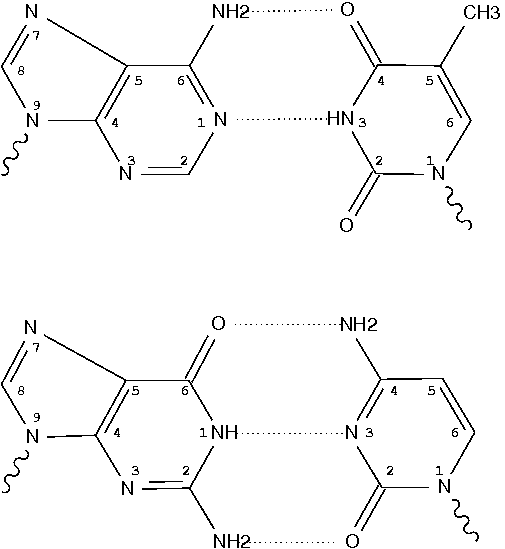
Without proper names for base ring atoms, DSSR is unable to identify nucleotides, resulting in the input structure being reported as 'no nucleotides found.' The same principle applies to amino acids in protein structures, such as specific naming conventions for amino nitrogen (N), carbonyl carbon (C), and alpha carbon (CA).
See also the blog posts "Mapping of modified nucleotides in DSSR" and "Name of base atoms in PDB formats".
Background and motivation
In late 2021, I came across the thread titled "create a 26 bp RNA from a 13 bp
system" on the PyMOL mailing list. The thread began with a user asking:
I have an RNA duplex with 13 base-pairs (attached). Is it possible to duplicate this system and then fuse the two molecules to create a 26 base-pair long system using the pymol.
The message is both concise and clear. The attached 13 base-pair RNA duplex (named model.pdb) makes the task easier to understand. An expert PyMOL user responded quickly, providing a set of suggested PyMOL commands along with warnings about the complexity of the task.
No, not automatically. Your RNA is very distorted from the standard A-form. I doubt any modeling program can accurately extend such a distorted helix. Maybe someone else will prove me wrong. ... You can align the terminal base pairs manually through a series of commands. If you try by dragging one copy relative to another, you will wind up pulling out all of your hair. The commands and patience will keep you out of the mad house.
DSSR offers unique capabilities to automatically manipulate nucleic acid structures. It also enables the duplication of an RNA duplex, as specifically requested by the original poster. In my initial response to the thread, I provided a DSSR-based solution for duplicating the RNA duplex without detailed explanations, aiming to confirm whether the result met the user's needs. The feedback was positive, as indicated below:
Thanks for proving me wrong. Congratulations on your duplicated model! Please share the commands that you used with DSSR to generate the duplicated helix. --- from the PyMOL responder
Thanks a lot for your help. The model you have duplicated is exactly what I am looking for (checked it with VMD). Unfortunately I do not have access to DSSR-Pro. Is there any way that I can reproduce your procedure with x3dna-dssr? I need to create different numbers of duplicates (2,4,6,5,8) for different systems and this will be very helpful. --- from the original poster
During that period (near the end of 2021), I was facing a funding gap. To address this challenge, we decided to license DSSR through Columbia Technology Ventures (CTV) and introduced a Pro version of DSSR for commercial users and academic institutions, providing advanced modeling features and dedicated support. Note that DSSR Pro Academic licenses entail a one-time fee of $1,020. The software can be installed on Windows, macOS, or Linux. While not explicitly included in the license agreement, I provide direct support to Pro license users via email, phone, or Zoom whatever convenient to help address their issues. I care about user experience, especially for those who invest in the Pro version.
Following user feedback, I shared detailed instructions on duplicating an RNA duplex using DSSR Pro. Gratefully, the original poster purchased a DSSR Pro Academic license and successfully duplicated the RNA helix. Later, we communicated via email to assist with other related tasks. This experience underscored the importance of engaging with the scientific community and addressing user needs to drive software development and adoption.
Detailed instructions
With funding from grant R24GM153869, I have transferred many DSSR Pro features into the free DSSR Academic version to better serve the scientific community. Included below are detailed step-by-step commands script for duplicating an RNA duplex using either DSSR Pro or the free DSSR Academic v2.5.2. The script runs instantaneously in a terminal window.
x3dna-dssr tasks -i=model.pdb --frame-pair=last -o=model1-ref-last.pdb
x3dna-dssr fiber --seq=GG --rna-duplex -o=conn.pdb
x3dna-dssr tasks -i=conn.pdb --frame-pair=first --remove-pair -o=ref-conn.pdb
x3dna-dssr tasks --merge-file='model1-ref-last.pdb ref-conn.pdb' -o=temp1.pdb
x3dna-dssr tasks -i=temp1.pdb --frame-pair=last --remove-pair -o=temp2.pdb
x3dna-dssr tasks -i=model.pdb --frame-pair=first -o=model1-ref-first.pdb
x3dna-dssr tasks --merge-file='temp2.pdb model1-ref-first.pdb' -o=duplicate-model.pdb
x3dna-dssr --order-residue -i=duplicate-model.pdb -o=temp3.pdb
x3dna-dssr --renumber-residue -i=temp3.pdb -o=temp4.pdb
x3dna-dssr --connect-file -i=temp4.pdb -o=RNA-duplicate.pdb
The procedure is essentially the same as the one used in "Building extended Z-DNA structures with backbones using DSSR". For completeness, I have included detailed
explanations for each step here as well.
-
Setting Up the Reference Frame:
- The first command places the 13 base-pair RNA duplex (
model.pdb) into the reference frame of its last base pair, resulting in model1-ref-last.pdb.
-
Creating the Fiber Connector:
- The
fiber model is constructed using an RNA duplex (--rna-duplex) with the sequence GG on the leading strand (conn.pdb). This connector is oriented into the reference frame of its first base pair.
- The first base pair is removed. Thus, the resulting coordinate file,
ref-conn.pdb, contains only one pair.
- Note: The sequence GG serves as a placeholder. It can be replaced with any other two bases: for instance, changing
--seq=GG to --seq=AA. Moreover, using --seq=GA10G allows for creating a linker with 10 adenines.
-
Merging PDB Files:
- The two PDB files,
model1-ref-last.pdb and ref-conn.pdb, share a common reference frame and are merged into a single file named temp1.pdb.
-
Adjusting the Reference Frame:
- The merged file (
temp1.pdb) is then aligned with the last base pair, which is subsequently removed to produce temp2.pdb. This completes the role of the GG fiber connector.
-
Reorienting the RNA Duplex:
- The original 13 base-pair RNA duplex (
model.pdb) is reoriented into the reference frame of its first base pair, generating model1-ref-first.pdb.
-
Final Merging:
- The two PDB files,
temp2.pdb and model1-ref-first.pdb, contain identical 13 base-pair RNA duplexes but in different orientations. They are merged into a single file (duplicate-model), establishing the final duplicated RNA structure.
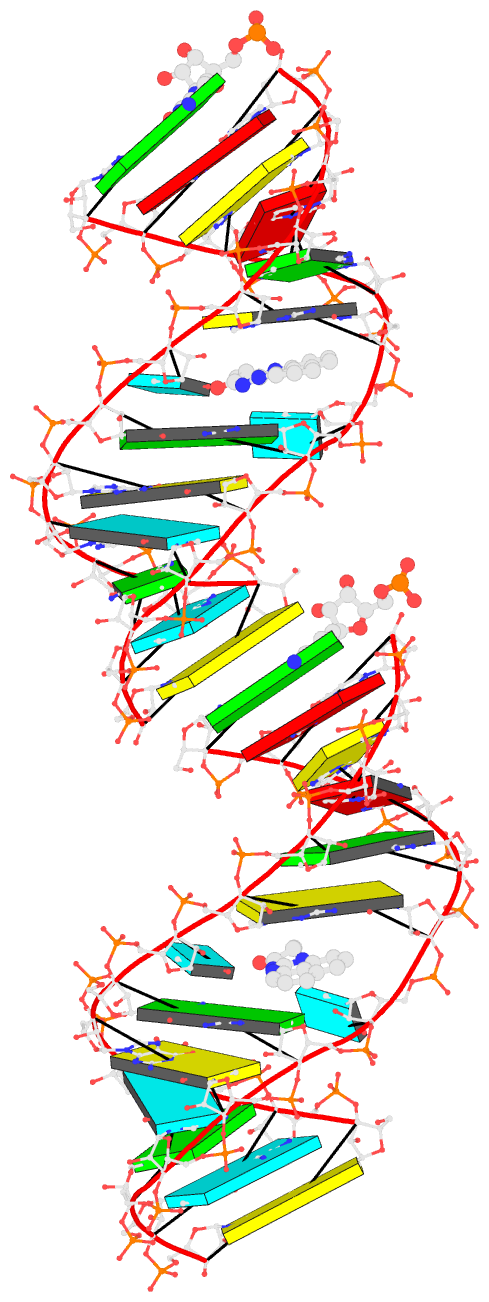
- Bookkeeping for Visualization:
The duplicated RNA helix is illustrated in the image below.
Some caveats
The original 13 base-pair RNA duplex (model.pdb) contains three main PDB format inconsistencies:
- Missing Chain Identifiers: The two strands lack proper chain identifiers in column 22.
- Incorrect Covalent Bond Distance: Nucleotides RU25 and RC26 are not covalently linked. Specifically, the distance between O3' of RU25 and P of RC26 is 3.5 Å, exceeding the expected 1.6 Å for a proper covalent bond.
- Misclassified Ligand Record: The ligand (LIG27) is incorrectly designated as
ATOM instead of the appropriate HETATM record.
A recent thread on the 3DNA Forum discussed 'Rebuilding Z-DNA' by extending an existing structure. The 3DNA rebuild program allows users to generate DNA or RNA structures with any user-specific sequence and corresponding base-pair/step parameters. This process is rigorous for atomic coordinates of base (and C1') atoms: running analyze on the rebuilt structure will yield the same set of parameters that users initially input. For more details, see the 2003 3DNA paper, the 2015 DSSR paper, and the DSSR User Manual.
The challenge lies in modeling the backbones. For right-handed A- or B-form DNA, users can build full-atomic models with canonical backbone conformations of C3'-endo or C2’-endo sugar conformations and anti glycosidic bonds. However, left-handed Z-DNA has unique structural features—such as syn-G, CpG, and GpC dinucleotides as building blocks instead of single nucleotides—that are not fully addressed by the 3DNA rebuild program.
DSSR (Pro version or the Academic v2.5.2) offers a solution by providing tools to build extended Z-DNA structures with proper backbones. The commands are as follows:
x3dna-dssr -i=1qbj.pdb1 --select-chains='D E' --delete-water -o=model.pdb
x3dna-dssr tasks -i=model.pdb --frame-pair=last -o=model1-ref-last.pdb
# poly d(GC) : poly d(GC)
x3dna-dssr fiber --z-dna --repeat=1 -o=conn.pdb
x3dna-dssr tasks -i=conn.pdb --frame-pair=first --remove-pair -o=ref-conn.pdb
x3dna-dssr tasks --merge-file='model1-ref-last.pdb ref-conn.pdb' -o=temp1.pdb
x3dna-dssr tasks -i=temp1.pdb --frame-pair=last --remove-pair -o=temp2.pdb
x3dna-dssr tasks -i=model.pdb --frame-pair=first -o=model1-ref-first.pdb
x3dna-dssr tasks --merge-file='temp2.pdb model1-ref-first.pdb' -o=duplicate-model.pdb
x3dna-dssr --order-residue -i=duplicate-model.pdb -o=temp3.pdb --po-bond=3.6
x3dna-dssr --renumber-residue -i=temp3.pdb -o=temp4.pdb
x3dna-dssr --connect-file -i=temp4.pdb -o=1qbj-duplicate.pdb --po-bond=3.6
The logic behind these commands is very straightforward, but technical details may look a bit complex for the uninitiated:
- The first command extracts the Z-DNA duplex consisting of chains D and E from PDB entry
1qbj.pdb1 (the first biological unit) and remove water molecules (model.pdb). The Z-DNA duplex has sequence: CGCGCG/CGCGCG.
- The next command sets the Z-DNA duplex (
model.pdb) into the reference frame of the last base pair, i.e., G-C (model1-ref-last.pdb).
- The
fiber model consists of the GpC dinucleotide step (conn.pdb), which is then set into the reference frame of the first base pair (G-C). The first G-C pair is removed from the coordinate file ref-conn.pdb which consists of only one C-G pair.
- The two PDB files,
model1-ref-last.pdb and ref-conn.pdb, share a common reference frame and are merged into a single PDB file (temp1.pdb).
- The merged PDB file (
temp1.pdb) is then set into the reference frame of last base pair(i.e., C-G) which is removed from the resulting coordinate file (temp2.pdb). Now the job of the GpC fiber connector is done.
- The Z-DNA duplex (
model.pdb) is once again set into the reference frame of the first base pair (i.e., C-G), leading to the coordinate file model1-ref-first.pdb.
- The two PDB files,
temp2.pdb and model1-ref-first.pdb, both consist of the same Z-DNA duplex but are in different orientations. They now share a common reference frame and are merged into the extended Z-DNA duplex (1qbj-duplicate.pdb).
- The last three commands (with options
--order-residue, --renumber-residue, --connect-file) are bookkeeping steps to ensure proper order and numbering of nucleotides along each chain, and generate the CONECT record for smooth view in PyMOL.
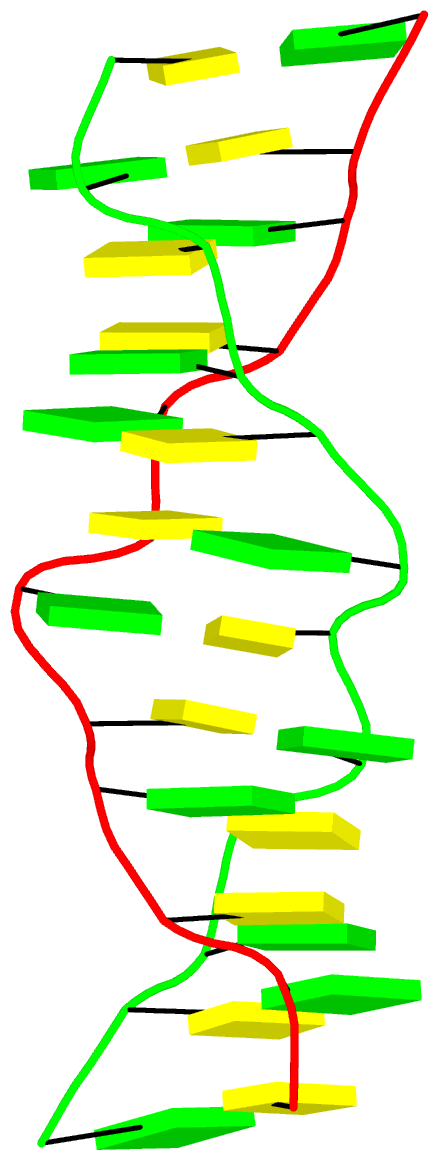
The final PDB coordinate file (1qbj-duplicate.pdb) can be downloaded, and visualized in DSSR-enabled cartoon-block representation as below:
In January 29, 2025, I received the following email request from a long-time DSSR user:
... recently noted that 3DNA/DSSR automatically maps non-standard nucleotides to standard nucleotides. I wonder if you would be willing to share with us your most current version of mappings?
I responded to the user the same day, with detailed information about the mapping process in DSSR. The user was happy with my response, and that thread was quickly closed with a positive note.
On April 22, 2025, a related question, titled "Can x3dna-dssr correctly handle N1-methyl-pseudouridine?", was asked on the 3DNA Forum. In answering the question on the Forum, I referred to my email response to the previous user.
I now realize that writing a detailed blog post explaining the mapping process would be beneficial for DSSR users. It would also enable me to easily reference this blog post in future interactions with users.
3DNA/DSSR performs automatic mapping of modified nucleotides (including pseudouridine) to their standard counterparts. Over the years, the method has proven to work well in real-world applications. It is one of the defining features that make DSSR just work. For example, for the tRNA 1ehz, DSSR automatically identifies the following 14 modified nucleotides (of 11 unique types):
# x3dna-dssr -i=1ehz.pdb
List of 11 types of 14 modified nucleotides
nt count list
1 1MA-a 1 A.1MA58
2 2MG-g 1 A.2MG10
3 5MC-c 2 A.5MC40,A.5MC49
4 5MU-t 1 A.5MU54
5 7MG-g 1 A.7MG46
6 H2U-u 2 A.H2U16,A.H2U17
7 M2G-g 1 A.M2G26
8 OMC-c 1 A.OMC32
9 OMG-g 1 A.OMG34
10 PSU-P 2 A.PSU39,A.PSU55
11 YYG-g 1 A.YYG37
Users could run DSSR on a set of structures of interest, and collect the unique mappings for a complete list of modified nucleotides.
Moreover, DSSR has the --nt-mapping option that allows users to control the mapping process. The screenshot below is taken from the relevant part of the DSSR manual.
For example, DSSR automatically maps 5MU (5-methyluridine 5′-monophosphate) to t (i.e., modified thymine) because of the 5-methyl group. With the option --nt-mapping='5MU:u', DSSR would take 5MU as a modified uracil. This option allows for multiple mappings separated by comma. The mapping of 5MU to u or t is obviously arbitrary. DSSR is robust against the ambiguity in designating a modified nucleotide to its nearest canonical counterpart. For example, mapping 5MU to u or t has minimal influence on DSSR-derived base-pair parameters and other structural features.

Background information on the mapping
Over the years, I've refined the heuristics of the mapping process. In the early days with 3DNA, I kept an ever increasing list in file baselist.dat with hundreds of entries like: MIA a that maps MIA as a modified A, denoted as lowercase a. In recent releases of DSSR, I keep only the standard ones, with a total of 48 entries like ADE A, and DG5 G etc. If a residue is not a standard one, the following C function is called to do the mapping. DSSR performs filtering to decide if a residue is a nucleotide, and if so R (purine) or Y (pyrimidine).
static void derive_new_nt_std_name(long resi, struct_mol *pdb, char *info)
{
char str[BUF512];
double d1 = DMAX, d2 = DMAX;
long C1_prime, N1, C5;
struct_residue *r = &pdb->residues[resi];
if (r->type[RESIDUE_NT_UNKNOWN]) {
sprintf(r->std_name, "__%c", Gvars.abasic);
return;
}
if (is_R(resi, pdb)) { /* purine */
if (residue_has_atom(" O6 ", resi, pdb)) /* with ' O6 ' */
strcpy(r->std_name, "__g");
else if (!residue_has_atom(" N6 ", resi, pdb) && /* no ' N6 ' but ' N2 ' */
residue_has_atom(" N2 ", resi, pdb))
strcpy(r->std_name, "__g");
else
strcpy(r->std_name, "__a");
} else { /* a pyrimidine */
if (residue_has_atom(" N4 ", resi, pdb))
strcpy(r->std_name, "__c");
else if (residue_has_atom(" C7 ", resi, pdb))
strcpy(r->std_name, "__t");
else
strcpy(r->std_name, "__u");
C1_prime = find_atom_in_residue(" C1'", resi, pdb);
N1 = find_atom_in_residue(" N1 ", resi, pdb);
if (atoms_same_model_chain_altloc(C1_prime, N1, pdb))
d1 = dist_atoms(C1_prime, N1, pdb);
if (!dval_in_range(d1, 1.0, 2.0)) {
C5 = find_atom_in_residue(" C5 ", resi, pdb);
if (atoms_same_model_chain_altloc(C1_prime, C5, pdb))
d2 = dist_atoms(C1_prime, C5, pdb);
if (dval_in_range(d2, 1.0, 2.0))
strcpy(r->std_name, "__p");
}
}
if (!Gvars.standalone) {
sprintf(str, "\n\tmatched nucleotide '%s' to '%c' for %s\n"
"\tverify and add an entry in <baselist.dat>\n",
r->res_name, r->std_name[2], info);
logit(str);
}
}
The legacy PDB format has a field called “altLoc” (alternate location indicator) for "ATOM/HETATM" records in the "Coordinate Section". The corresponding documentation is excerpted below:
COLUMNS DATA TYPE FIELD DEFINITION
-----------------------------------------------------------------------
17 Character altLoc Alternate location indicator.
-
AltLoc is the place holder to indicate alternate conformation. The alternate conformation can be in the entire polymer chain, or several residues or partial residue (several atoms within one residue). If an atom is provided in more than one position, then a non-blank alternate location indicator must be used for each of the atomic positions. Within a residue, all atoms that are associated with each other in a given conformation are assigned the same alternate position indicator. There are two ways of representing alternate conformation- either at atom level or at residue level (see examples).
- For atoms that are in alternate sites indicated by the alternate site indicator, sorting of atoms in the ATOM/HETATM list uses the following general rules:
- In the simple case that involves a few atoms or a few residues with alternate sites, the coordinates occur one after the other in the entry.
- In the case of a large heterogen groups which are disordered, the atoms for each conformer are listed together.
In mmCIF format, AltLoc is under the atom_site category, with attribute name label_alt_id: i.e., labelled as _atom_site.label_alt_id. It is a required data item and appears in 43% of entries in the PDB.
In 3DNA and DSSR, AltLoc has a default value of "A1 ": an atom is taken into consideration if its AltLoc field (a single character) is space, A, or 1, otherwise it is ignored. Note that for mmCIF format, AltLoc field with dot (.) or question mark (?) is taken as space. Customized AltLoc values can be set via the --altloc option in DSSR.
Here is an example. PDB entry 7o1h is a 31-mer synthetic construct, with a hybrid-2R quadruplex-duplex of 3(-P-P-Lw) topology and three syn guanosines. It contains two modified residues designated BGM (BGM26 and BGM28), 8-bromo-2'-deoxyguanosine-5'-monophosphate, with AltLoc set to B. By default, DSSR detects only one G-tetrad, consisting of DG5,DG9,DG13,DG27, ignoring the two G-tetrads with BGM26 and BGM28. With the --altloc=B option (space is always included), all three G-tetrads are detected and the G-quadruplex (a G4-stem) is then automatically annotated as 3(-P-P-Lw):
# x3dna-dssr -i=7o1h-assembly1.cif --altloc=B
List of 2 types of 3 modified nucleotides
nt count list
1 BGM-g 2 A.BGM26,A.BGM28
2 THM-t 1 A.THM1
List of 1 G4-stem
Note: a G4-stem is defined as a G4-helix with backbone connectivity.
Bulges are also allowed along each of the four strands.
stem#1[#1] layers=3 INTRA-molecular loops=3 descriptor=3(-P-P-Lw) note=hybrid-2R(3+1) UUUD hybrid-(mixed)
1 glyco-bond=---s sugar=---. groove=--wn WC-->Major nts=4 GGGg A.DG4,A.DG8,A.DG12,A.BGM28
2 glyco-bond=---s sugar=--.- groove=--wn WC-->Major nts=4 GGGG A.DG5,A.DG9,A.DG13,A.DG27
3 glyco-bond=---s sugar=---- groove=--wn WC-->Major nts=4 GGGg A.DG6,A.DG10,A.DG14,A.BGM26
step#1 pm(>>,forward) area=13.57 rise=3.39 twist=26.7
step#2 pm(>>,forward) area=12.00 rise=3.44 twist=28.4
strand#1 U DNA glyco-bond=--- sugar=--- nts=3 GGG A.DG4,A.DG5,A.DG6
strand#2 U DNA glyco-bond=--- sugar=--- nts=3 GGG A.DG8,A.DG9,A.DG10
strand#3 U DNA glyco-bond=--- sugar=-.- nts=3 GGG A.DG12,A.DG13,A.DG14
strand#4 D DNA glyco-bond=sss sugar=.-- nts=3 gGg A.BGM28,A.DG27,A.BGM26
loop#1 type=propeller strands=[#1,#2] nts=1 T A.DT7
loop#2 type=propeller strands=[#2,#3] nts=1 T A.DT11
loop#3 type=lateral strands=[#3,#4] nts=11 ACGCGCAGCGT A.DA15,A.DC16,A.DG17,A.DC18,A.DG19,A.DC20,A.DA21,A.DG22,A.DC23,A.DG24,A.DT25
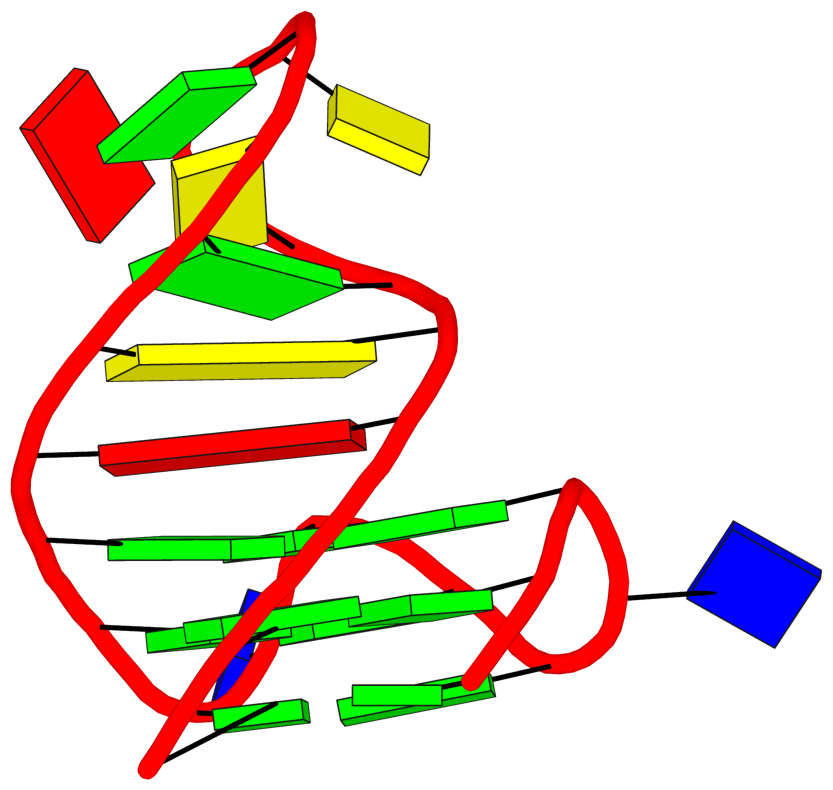
See G4.x3dna.org for DSSR-enabled annotation and visualization of this G4 structure. Here is the G4-stem in the frame of reference of 5' DG4 (bottom right), following the convention of Dvorkin et al. (2018). It is orientated automatically based on the standard base-reference frame (Olson et al. (2001)) of DG4.
References:
- Dvorkin, Scarlett A., Andreas I. Karsisiotis, and Mateus Webba Da Silva. 2018. “Encoding Canonical DNA Quadruplex Structure.” Science Advances 4 (8): eaat3007. https://doi.org/10.1126/sciadv.aat3007.
- Olson, Wilma K, Manju Bansal, Stephen K Burley, Richard E Dickerson, Mark Gerstein, Stephen C Harvey, Udo Heinemann, et al. 2001. “A Standard Reference Frame for the Description of Nucleic Acid Base-Pair Geometry.” Journal of Molecular Biology 313 (1): 229–37. https://doi.org/10.1006/jmbi.2001.4987.