Announcing wDSSR: The Next-Generation Web Interface to X3DNA-DSSR
Dear 3DNA/DSSR Community,
We are thrilled to announce the official launch of wDSSR (https://web.x3dna-dssr.org/), the powerful new web interface to the X3DNA-DSSR analytical engine.
Developed by Drs. Shuxiang Li and Xiang-Jun Lu and supported by NIH grant R24GM153869, wDSSR represents a major leap forward from our highly popular 2019 Web 3DNA 2.0 framework. While Web 3DNA 2.0 has faithfully served the community for the analysis, visualization, and modeling of 3D nucleic acid structures, wDSSR was built from the ground up to take full advantage of modern web technologies and the latest DSSR backend capabilities.
A Modern, Streamlined Scientific Workflow
We have completely overhauled the user interface to provide a clean, intuitive, and task-driven experience. The core modeling and analysis tools are now seamlessly organized into a logical, single-word scientific workflow: Analyze, Rebuild, Model, Circularize, Mutate, Assemble, and Visualize.
Spotlight Feature: The "Assemble" Module
One of the most exciting upgrades is the newly renamed Assemble tab (formerly "Composite"). This advanced composite model builder allows you to effortlessly construct complex, higher-order models by linking any combination of nucleic acid duplexes or protein-DNA/RNA complexes. You can quickly connect up to six distinct target structures, ranging from simple linked A-DNA and B-DNA duplexes to large, protein-decorated structural assemblies.
Immediate Global Adoption
Although wDSSR has just launched, we are incredibly humbled to share that it is already seeing rapid worldwide adoption! According to recent network infrastructure data, the new interface is actively being used by researchers across North America, South America, Europe, and Asia. Within just a few days, we have recorded active sessions from prestigious institutions around the globe, including:
- The Weizmann Institute of Science in Israel
- Katholieke Universiteit Leuven in Belgium
- Queen's University in Canada
- Universidad Nacional Autonoma de Mexico (UNAM) in Mexico
- Emory University and the Wadsworth Centers Laboratories and Research in the United States
- Jawaharlal Nehru University and the China Education and Research Network in Asia
How to Cite
While a dedicated paper for wDSSR is currently in preparation, researchers should cite the server using its URL (https://web.x3dna-dssr.org/) alongside the 2019 Web 3DNA 2.0 paper and the foundational 2015 DSSR paper. Full details and funding acknowledgements can be found on our newly consolidated About page.
We invite you all to try out the new wDSSR platform! As always, your feedback is invaluable to us, and we encourage you to share your thoughts, questions, and structural models via the newly updated Questions & Feedback link in the wDSSR footer.
Happy modeling!
By following DSSR citations, I recently noticed a bioRxiv preprint, titled "Assessment of nucleic acid structure prediction in CASP16" by Kretsch et al. The portion where DSSR is mentioned is as follows:
Secondary structures were extracted from CASP16 models with DSSR (v1.9.9-2020feb06). Some models, in particular due to large clashes, failed to run (Supplemental Table 1). The base-pair list was extracted from the table in the output file directly because the dot-bracket structure produced by DSSR, in particular for multimers, can contain errors.
While pleased to see DSSR cited in this significant study, I am concerned about the reported issues and would like to investigate the specific structures and error messages encountered. To better understand the problems and potentially find solutions, I have reached out to the authors for further details. Here is the message I sent initially:
You said DSSR failed to run on some models with large clashes. Could you please share the specific models and the error messages you encountered? I would also be interested in seeing the exact errors you observed in the DSSR-derived DBN for multi-mers. It would be a great opportunity for me to improve DSSR in this area, which would benefit both your group and the broader community. If you are willing to share them, please provide details—preferably on the **public 3DNA Forum**. Don’t hesitate to share openly any bugs or limitations you’ve encountered with DSSR.
The authors responded promptly and provided detailed information about the specific models and error messages encountered. After several
iterations, I successfully resolved the issues and released an updated version of DSSR, namely v2.5.4-2025jun04. You can find the release notes
here. This experience underscores the importance of proactively engaging with the community to enhance the functionality and reliability of a software tool.
In this blog post, I aim to share the specifics of these issues and the steps taken to address them. For ease of reading, I have formatted the response/feedback from the authors in red block quotes, and my enquiries/comments in blue. The beginning round of correspondence is as below.
Do note, the predictors in casp submit some truly atrocious models --- eg 14 atoms all at the exact same x-y-z coordinate. These errors would be with his v1.9.9-2020feb06 install though not your latest version. Would you still like them?
Yes, I would like to see how DSSR behaves with these models. Ideally, it should not crash, but output some warning messages. Only through such testing can we improve the robustness of DSSR. Overall, the more feedback I get, the better.
Buffer overflow bug in DSSR
Most of errors I had with dssr were due to clashes and all zero xyz predictions by predictors, for all of which dssr did not give an error message when dssr failed. There was a case where the prediction looked reasonable but dssr failed with the error message `dssr error*** buffer overflow detected ***`. Please see attached for the 2 pdbs that gave this error.
The two PDB files I received were R1283v3TS294_1o and R1283v3TS294_2o, as listed in Supplementary Table 1: "List of unscored models," with the "Reasons" column indicating a dssr error*** buffer overflow detected ***. I immediately acknowledged receipt of these files, as shown in the following message:
Thank you for sending me the two PDB files which caused DSSR to fail. I can verify the issue and will try to fix the bug ASAP. I'll keep you posted.
Using these data files, I was able to quickly fix the buffer overflow bug. The following is my response to the authors within one day after receiving the files:
With your sample PDB files, I have traced the issue that caused DSSR to fail. The bug was due to a 53-way (`R1283v3TS294_1o`) and 40-way (`R1283v3TS294_2o`) junction loops which are far from the norm. DSSR sets a default limit for the summary line for each loop which is more than sufficient for all normal PDB entries, but falls short for these unusual cases, leading to out of array boundaries. See the attached DSSR output after the bug fix for more details.
This is a clear example where user feedback is crucial for improving the software, which makes it better serve the community.
Zero xyz coordinates and large clashes
After fixing the out-of-bound bug, I also requested other problematic predicted models from the authors, as shown in the following message:
Along the line, please provide the sample PDB files:
- with zero xyz predictions -- I am curious to see what it looks like.
- where the DSSR-derived DBN is problematic for multi-mers
After solving these issues, I will release a new version of DSSR that would make your analysis more straightforward, and benefit other users as well.
The authors responded with the following message:
Thanks for looking into this. Here are some more examples with superimposed structures, large clash, and all zero xyzs in the zip file.
The ZIP file (error_examples.zip) contains three folders (all_zero_xyz, clash and superimposed), each with some problematic models in PDB format. Once again, I promptly acknowledged receipt of the files and was able to reproduce the reported issues.
Garbage in, garbage out. Given these problematic models, one should not expect DSSR to extract any meaningful information from them. Nonetheless, I am committed to enhancing the software so that it can handle such cases more effectively by providing clear error messages and terminating gracefully rather than crashing.
After several days of thinking, elaboration, intensive coding, and testing, I solved the problems. I then communicated the results to the authors in the following detailed message:
Thanks for the sample PDB files (`error_examples`) with all zero XYZ coordinates, large clashes, and superimposed structures. They helped me to understand the issues, think in context, and find solutions. Let's look them one by one:
1. `all_zero_xyz`: These two files `R1211TS159_1` and `R1211TS159_2` have identical contents, except for the MODEL IDs (1 and 2, respectively). Atoms with all-zero XYZ coordinates are a special case of duplicated coordinates. This has led me to implement a check for duplicated coordinates in an input file. The revised DSSR now reports duplicated coordinates and their corresponding atoms, and it quits if the number of duplicated atoms exceeds a certain threshold. For `R1211TS159_1`, the revised DSSR output would be as below:
1 [e] xyz repeated 1904 times:[0.000 0.000 0.000] 1509-P@0.G1 3412-C6@0.C90
[w] no-of-repeats=1 max-freq=1904
...too many duplicates... quit!
2. `clash`: Both files `R1250TS208_1o` and `R1250TS417_1o` contain multiple models, as visible in PyMOL. Each PDB file uses a single MODEL/END pair to include all its models. This setup is akin to an NMR ensemble but without MODEL/ENDMDL delimiters, which leads to clashes when analyzed together. I have revised DSSR to explicitly check for such clashes and terminate execution if too many are detected. Using `R1250TS208_1o` as an example, the DSSR output would be as below:
[i] 0.G1 and 1.G1 in clashes: min_dist=0.57
[i] 0.G1 and 3.G1 in clashes: min_dist=0.35
[i] 0.G1 and 4.G1 in clashes: min_dist=0.41
...too many clashes... quit!
The above list contains only three of the many clashes detected in this file. One can notice immediately the G1 nucleotides from chains `0`, `1`, `3`, and `4` are in clashes (see the attached file `clashes_208.pdb`, which contains only G1 nucleotides from the four chains).
3. `superimposed`: The five example files (`R1283v3TS304_1o` ... `R1283v3TS304_5o`) have similar issues as the clash cases. Running the revised DSSR on `R1283v3TS304_1o` would produce the following output:
[i] 0.A1 and 2.A1 in clashes: min_dist=0.74
[i] 0.A1 and 3.A1 in clashes: min_dist=0.78
[i] 0.A1 and 4.A1 in clashes: min_dist=0.56
...too many clashes... quit!
Here A1 nucleotides from chains `0`, `2`, `3` , and `4` are in clashes (see the attached `superimpose-1.pdb`).
How the `clash` and `superimposed` categories are supposed to be different? They look similar to me.
Overall, the `error_examples` (in `all_zero_xyz`, `clash`, and `superimposed`) pose problems because they do not contain valid DNA/RNA structures as a whole. DSSR cannot extract meaningful information from these files. However, the revised DSSR explicitly highlights these issues, saving users from spending time on invalid data. Do these DSSR revisions make sense to you?
In the end, I am glad to receive the following feedback from the authors:
Thanks, these revisions all make sense! The examples I sent on clashes and superimposed were actually similar and I think the error output makes sense as well.
Final thoughts
This blog post offers an in-depth look at my efforts to enhance DSSR. As the developer of this software product, I am deeply committed to ensuring its quality and usability. I extend my gratitude to the authors for their valuable feedback and assistance in resolving these issues. In return, the updated version of DSSR (v2.5.4-2025jun04) should not only streamline their workflow but also benefit the broader user community.
For those who read through this lengthy post, I want to emphasize that DSSR is actively supported: I am here to listen and help. Any questions related to its use, bug reports, or feature requests are warmly welcomed on the 3DNA Forum. As I’ve mentioned before, please don’t hesitate to share any negative experiences or bugs with DSSR—just ensure to provide specific details so others can reproduce the issue. I will address these concerns as soon as I’m aware of them and will frankly acknowledge any mistakes I may have made. My goal is for DSSR to be a reliable software tool that the community can trust and build upon.
References
Kretsch,R.C. et al. (2025) Assessment of nucleic acid structure prediction in CASP16. bioRxiv; https://doi.org/10.1101/2025.05.06.652459.

In DSSR, the --frame option allows users to reorient a nucleic acid structure using the standard base reference frame (see Olson et al., 2001). This option can be applied not only to an individual base frame but also a base-pair frame, or the middle frame between two bases or base pairs. These variations facilitate the alignment of nucleic acid structures for a wide range of comparative analyses. In this blog post, I will demonstrate how to use the --frame option with concrete examples, enabling readers to apply this unique DSSR feature to their own projects.
The standard base reference frame
The standard base reference frame is derived from an idealized Watson-Crick base pairing geometry (top-left, figure below). The x-axis points in the direction of the major groove along what would be its pseudo-dyad axis—that is, the perpendicular bisector of the C1'...C1' vector spanning the base pair. The y-axis runs along the long axis of the idealized base-pair in the direction of the sequence strand, parallel to the C1'...C1' vector, and is displaced so as to pass through the intersection between the (pseudo-dyad) x-axis and the vector connecting the pyrimidine Y(C6) and purine R(C8) atoms. The z-axis is defined by the right-handed rule. For right-handed A- and B-DNA, the z-axis accordingly points along the 5' to 3' direction of the sequence strand.
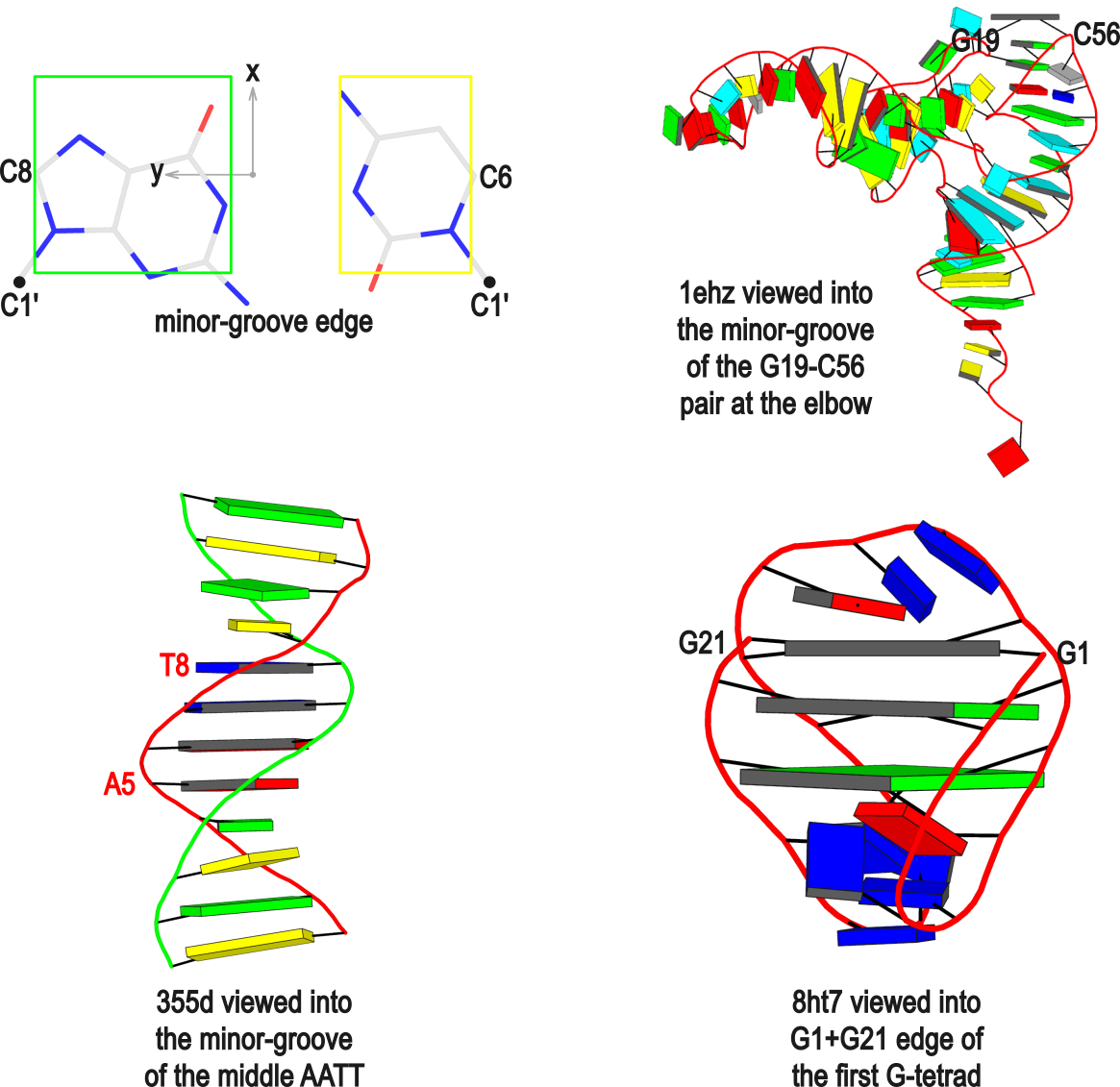
Typical usages of the --frame option
Using the classic B-DNA dodecamer PDB entry 355d as an example, DSSR can be run with the --frame option as follows:
# 1...5..8....
# chain A: 5'-CGCGAATTCGCG -3'
# chain B: 3'-GCGCTTAAGCGC -5'
# reorient 355d in the reference frame of C1 on chain A
x3dna-dssr -i=355d.pdb --frame=A.1 -o=355d-b1.pdb
# reorient 355d in the frame of the Watson-Crick pair C1-G24
x3dna-dssr -i=355d.pdb --frame=A.1:wc -o=355d-bp1.pdb
# ... with the minor-groove of pair C1-G24 facing the viewer
x3dna-dssr -i=355d.pdb --frame=A.1:wc-minor -o=355d-bp1-minor.pdb
# with the minor-groove of the middle AATT tract facing the viewer
x3dna-dssr -i=355d.pdb --frame='A.5:wc-minor A.8:wc' -o=355d-AATT-minor.pdb
# Rendered in cartoon-blocks with base-pair blocks, and black minor-groove
# Load 355d-AATT-minor.pml into PyMOL (bottom-left, figure above)
x3dna-dssr -i=355d-AATT-minor.pdb --cartoon-block --block-file=wc-minor -o=355d-AATT-minor.pml
The abbreviated notation A.1 refers to nucleotide numbered 1 (as indicated in the coordinates file) on chain A. Here, it denotes C1, as shown at the top of the listing. Similarly, A.5 and A.8 correspond to nucleotides A5 and T8 on chain A, respectively. In most cases, such as with 355d, the combination of chain identifier and residue number is sufficient to uniquely identify a nucleotide. More generally, other information such as model number or insertion code may be needed to specify a particular nucleotide.
In the above listing, wc after the colon (for example, A.1:wc) specifies the Watson-Crick base pair that the corresponding nucleotide participates in. Meanwhile, minor transforms the structure so that the minor-groove of the base (or base pair, or step) faces the viewer. The keywords wc and minor are settings that influence the construction or view of the frame. Case or order does not matter for these keywords as long as there is a match—for example, minor+wc works the same as wc-minor.
Two other examples combining the --frame option with cartoon-block representations
The intuitive geometric meaning of the standard base reference frame combined with the DSSR-enabled cartoon-block representation allows for an enhanced understanding of intricate structural features. In the top-right panel of the figure above, we see the classic yeast phenylalanine tRNA (PDB entry 1ehz) viewed into the minor-groove of the pseudo-knotted G19-C56 pair at the elbow of the L-shaped tertiary structure. The stacking interactions of the purines at the top-right of the panel are clearly visible in this view. In the bottom-right panel, an anti-parallel G-quadruplex from PDB entry 8ht7 is shown. The G-tetrads are automatically identified and rendered as square blocks, all with DSSR. This representation makes the chair conformation of the three-layered anti-parallel G-quadruplex crystal clear. The DSSR commands used are listed below:
# yeast tRNA (1ehz)
x3dna-dssr -i=1ehz.pdb --frame=A.19:wc-minor -o=1ehz-elbow.pdb
x3dna-dssr -i=1ehz-elbow.pdb --cartoon-block --block-file=wc-minor -o=1ehz-elbow.pml
# anti-parallel chair-shaped G-quadruplex (8ht7)
x3dna-dssr -i=8ht7.pdb --select=nts -o=8ht7-nts.pdb # extract nucleotides, ignore amino acids
# reorient 8ht7 in the frame of the G-tetrad involving G1, in edge view
x3dna-dssr -i=8ht7-nts.pdb --frame=A.1:G4-minor -o=8ht7-Gtetrad.pdb
x3dna-dssr -i=8ht7-Gtetrad.pdb --block-cartoon --block-file=G4-minor -o=8ht7-Gtetrad.pml
References
Olson,W.K. et al. (2001) A standard reference frame for the description of nucleic acid base-pair geometry. Journal of Molecular Biology, 313, 229–237.
The 3DNA suite includes the mutate_bases program, which, as its name suggests, mutates bases while maintaining the backbone conformation. This feature was incorporated into the suite following user feedback and has been utilized in several studies before being formally published in the Li et al. (2019) paper. A key advantage is that the mutation process preserves both the geometry of the sugar-phosphate backbone and the base reference frame, encompassing position and orientation. Consequently, re-analyzing the mutated model yields identical base-pair and step
parameters as those of the original structure.
In DSSR, the standalone mutate_bases program has become the mutate sub-command with enhanced functionality and improved usability, as documented in the User Manual. The mutate module allows users to perform base mutations efficiently and effectively by taking advantage of the powerful DSSR analysis engine.
To further expand the modeling capabilities of the DSSR, v2.5.3 introduced the --mutate-type option to allow for backbone mutations, based on the base reference frame. Furthermore, the target can be any fragment, regardless of length or composition, rather than just a single nucleotide. When combined with the rebuild module, this feature significantly enhances DSSR’s ability to model nucleic acid structures.
Here is an example of modeling PDB entry 1msy, a 27-nt structure (1msy.pdb) that mimics the sarcin/ricin loop from E. coli 23S ribosomal RNA.
x3dna-dssr analyze -i=1msy.pdb --ss --rebuild -o=1msy-expt.out
mv dssr-ssStepPars.txt 1msy-step.txt
x3dna-dssr rebuild --backbone=RNA --par-file=1msy-step.txt -o=1msy-step.pdb
x3dna-dssr -i=1msy.pdb --select-resi='A 2654' -o=1msy-A2654.pdb
x3dna-dssr -i=1msy.pdb --select-resi='A 2655' -o=1msy-G2655.pdb
x3dna-dssr -i=1msy-A2654.pdb --frame=2654 -o=frame___A.pdb
x3dna-dssr -i=1msy-G2655.pdb --frame=2655 -o=frame___G.pdb
x3dna-dssr mutate -i=1msy-step.pdb --entry='num=8 to=A; num=9 to=G' -o=1msy-C2endo.pdb --mutate-part=whole
x3dna-dssr --connect-file -i=1msy-C2endo.pdb -o=1msy-C2endo-cnt.pdb --po-bond=5.0
- The
analyze step uses options --ss and --rebuild to generate the file dssr-ssStepPars.txt (containing base-step parameters), which is then renamed to 1msy-step.txt. The rebuild step employs 1msy-step.txt to construct a structure (1msy-step.pdb) with regular C3'-endo sugar RNA backbone conformation. Note that the rebuilt structure has nucleotides numbered from 1 to 27, while in the PDB 1msy, they correspond to 2647 to 2673, respectively.
- However, the A2654 and G2655 dinucleotides in 1msy are actually in C2'-endo sugar conformation, creating the S-shaped structure around the GpU platform. The above rebuilt structure does not reflect this distortion. So we extract A2654 and G2655 with
--select-resi and then put each in its standard base reference frame, named frame___A.pdb and frame___G.pdb, respectively.
- Now we mutate A8 and G9 in the rebuilt structure
1msy-step.pdb to A and G with option --mutate-part=backbone to ensure the backbone conformations are changed according to those in frame___A.pdb and frame___G.pdb, respectively. The resulting structure is named 1msy-C2endo.pdb. Now the S-shape around the GpU platform is preserved, even though the backbone are not always covalently connected, due to large O3'(i-1) to P(i) distances between neighboring nucleotides. The last step is to generate CONECT records with --connect-file option to connect the backbone atoms explicitly, resulting in more smooth backbone cartoon representation in PyMOL as shown below.
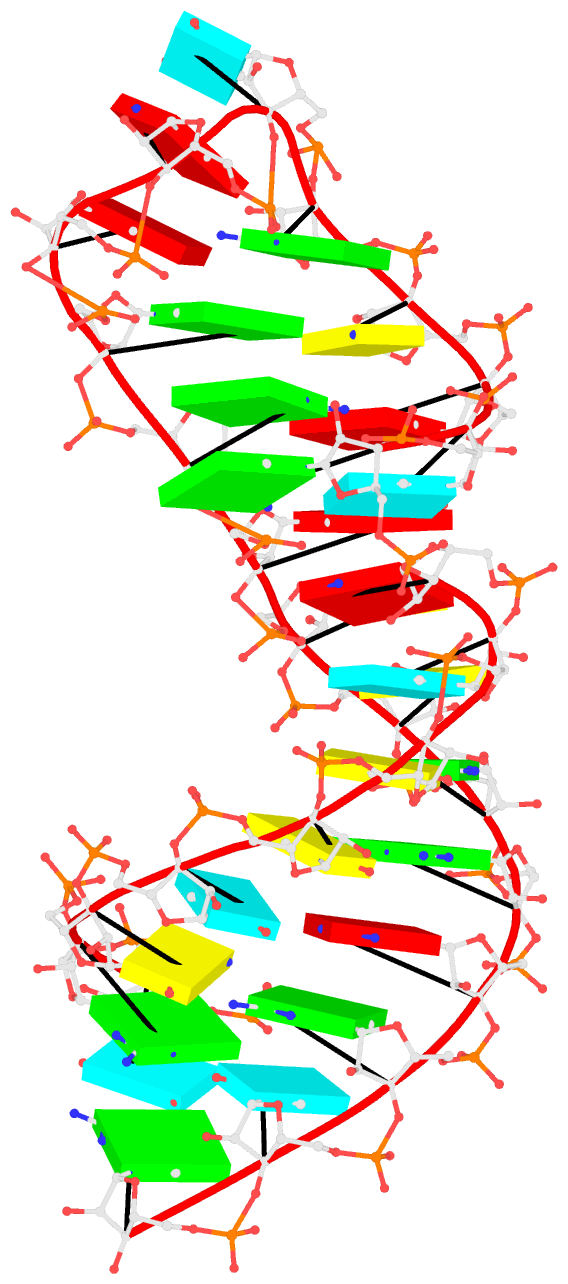
As noted in the Li et al. (2019) paper, users can optimize this approximate backbone connection using Phenix, while keeping the base atoms fixed. The 3DNA-Phenix combination leads to a model where the base geometry strictly follows the parameters prescribed in the user-specified file, and the backbone is regularized with improved stereochemistry and a ‘smooth’ appearance in ribbon representation.
There are other variants of the DSSR mutate module, including for building Z-DNA backbones. However, the above example is sufficient to demonstrate the power of the integrated approach enabled by DSSR for the analysis and modeling of nucleic acid structures. See the DSSR User Manual for more details.
References
Li,S. et al. (2019) Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Nucleic Acids Res., 47, W26–W34.
Background and motivation
In late 2021, I came across the thread titled "create a 26 bp RNA from a 13 bp
system" on the PyMOL mailing list. The thread began with a user asking:
I have an RNA duplex with 13 base-pairs (attached). Is it possible to duplicate this system and then fuse the two molecules to create a 26 base-pair long system using the pymol.
The message is both concise and clear. The attached 13 base-pair RNA duplex (named model.pdb) makes the task easier to understand. An expert PyMOL user responded quickly, providing a set of suggested PyMOL commands along with warnings about the complexity of the task.
No, not automatically. Your RNA is very distorted from the standard A-form. I doubt any modeling program can accurately extend such a distorted helix. Maybe someone else will prove me wrong. ... You can align the terminal base pairs manually through a series of commands. If you try by dragging one copy relative to another, you will wind up pulling out all of your hair. The commands and patience will keep you out of the mad house.
DSSR offers unique capabilities to automatically manipulate nucleic acid structures. It also enables the duplication of an RNA duplex, as specifically requested by the original poster. In my initial response to the thread, I provided a DSSR-based solution for duplicating the RNA duplex without detailed explanations, aiming to confirm whether the result met the user's needs. The feedback was positive, as indicated below:
Thanks for proving me wrong. Congratulations on your duplicated model! Please share the commands that you used with DSSR to generate the duplicated helix. --- from the PyMOL responder
Thanks a lot for your help. The model you have duplicated is exactly what I am looking for (checked it with VMD). Unfortunately I do not have access to DSSR-Pro. Is there any way that I can reproduce your procedure with x3dna-dssr? I need to create different numbers of duplicates (2,4,6,5,8) for different systems and this will be very helpful. --- from the original poster
During that period (near the end of 2021), I was facing a funding gap. To address this challenge, we decided to license DSSR through Columbia Technology Ventures (CTV) and introduced a Pro version of DSSR for commercial users and academic institutions, providing advanced modeling features and dedicated support. Note that DSSR Pro Academic licenses entail a one-time fee of $1,020. The software can be installed on Windows, macOS, or Linux. While not explicitly included in the license agreement, I provide direct support to Pro license users via email, phone, or Zoom whatever convenient to help address their issues. I care about user experience, especially for those who invest in the Pro version.
Following user feedback, I shared detailed instructions on duplicating an RNA duplex using DSSR Pro. Gratefully, the original poster purchased a DSSR Pro Academic license and successfully duplicated the RNA helix. Later, we communicated via email to assist with other related tasks. This experience underscored the importance of engaging with the scientific community and addressing user needs to drive software development and adoption.
Detailed instructions
With funding from grant R24GM153869, I have transferred many DSSR Pro features into the free DSSR Academic version to better serve the scientific community. Included below are detailed step-by-step commands script for duplicating an RNA duplex using either DSSR Pro or the free DSSR Academic v2.5.2. The script runs instantaneously in a terminal window.
x3dna-dssr tasks -i=model.pdb --frame-pair=last -o=model1-ref-last.pdb
x3dna-dssr fiber --seq=GG --rna-duplex -o=conn.pdb
x3dna-dssr tasks -i=conn.pdb --frame-pair=first --remove-pair -o=ref-conn.pdb
x3dna-dssr tasks --merge-file='model1-ref-last.pdb ref-conn.pdb' -o=temp1.pdb
x3dna-dssr tasks -i=temp1.pdb --frame-pair=last --remove-pair -o=temp2.pdb
x3dna-dssr tasks -i=model.pdb --frame-pair=first -o=model1-ref-first.pdb
x3dna-dssr tasks --merge-file='temp2.pdb model1-ref-first.pdb' -o=duplicate-model.pdb
x3dna-dssr --order-residue -i=duplicate-model.pdb -o=temp3.pdb
x3dna-dssr --renumber-residue -i=temp3.pdb -o=temp4.pdb
x3dna-dssr --connect-file -i=temp4.pdb -o=RNA-duplicate.pdb
The procedure is essentially the same as the one used in "Building extended Z-DNA structures with backbones using DSSR". For completeness, I have included detailed
explanations for each step here as well.
-
Setting Up the Reference Frame:
- The first command places the 13 base-pair RNA duplex (
model.pdb) into the reference frame of its last base pair, resulting in model1-ref-last.pdb.
-
Creating the Fiber Connector:
- The
fiber model is constructed using an RNA duplex (--rna-duplex) with the sequence GG on the leading strand (conn.pdb). This connector is oriented into the reference frame of its first base pair.
- The first base pair is removed. Thus, the resulting coordinate file,
ref-conn.pdb, contains only one pair.
- Note: The sequence GG serves as a placeholder. It can be replaced with any other two bases: for instance, changing
--seq=GG to --seq=AA. Moreover, using --seq=GA10G allows for creating a linker with 10 adenines.
-
Merging PDB Files:
- The two PDB files,
model1-ref-last.pdb and ref-conn.pdb, share a common reference frame and are merged into a single file named temp1.pdb.
-
Adjusting the Reference Frame:
- The merged file (
temp1.pdb) is then aligned with the last base pair, which is subsequently removed to produce temp2.pdb. This completes the role of the GG fiber connector.
-
Reorienting the RNA Duplex:
- The original 13 base-pair RNA duplex (
model.pdb) is reoriented into the reference frame of its first base pair, generating model1-ref-first.pdb.
-
Final Merging:
- The two PDB files,
temp2.pdb and model1-ref-first.pdb, contain identical 13 base-pair RNA duplexes but in different orientations. They are merged into a single file (duplicate-model), establishing the final duplicated RNA structure.
- Bookkeeping for Visualization:
The duplicated RNA helix is illustrated in the image below.
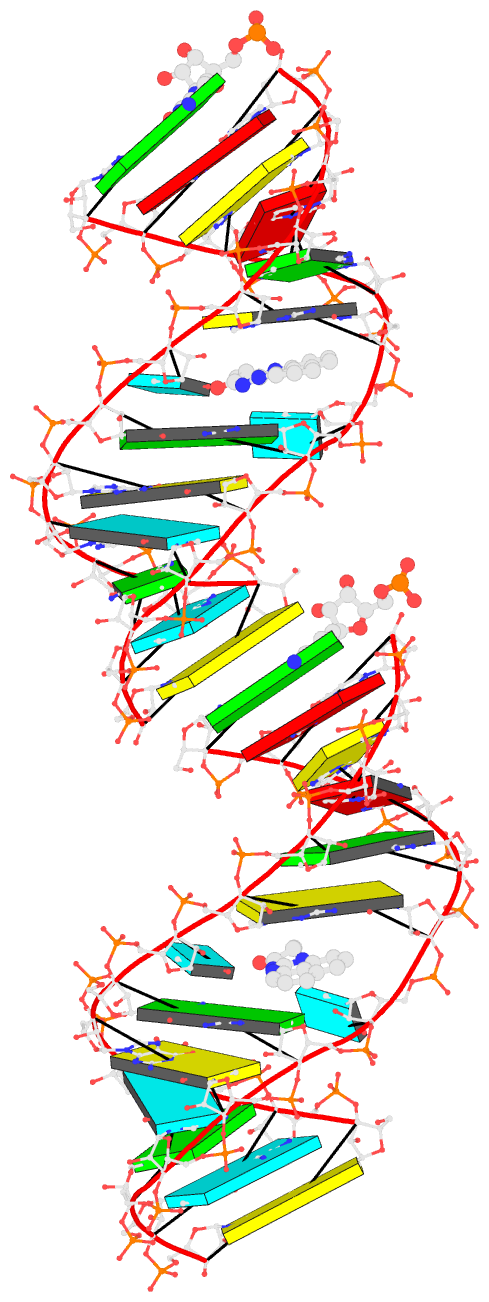
Some caveats
The original 13 base-pair RNA duplex (model.pdb) contains three main PDB format inconsistencies:
- Missing Chain Identifiers: The two strands lack proper chain identifiers in column 22.
- Incorrect Covalent Bond Distance: Nucleotides RU25 and RC26 are not covalently linked. Specifically, the distance between O3' of RU25 and P of RC26 is 3.5 Å, exceeding the expected 1.6 Å for a proper covalent bond.
- Misclassified Ligand Record: The ligand (LIG27) is incorrectly designated as
ATOM instead of the appropriate HETATM record.
A recent thread on the 3DNA Forum discussed 'Rebuilding Z-DNA' by extending an existing structure. The 3DNA rebuild program allows users to generate DNA or RNA structures with any user-specific sequence and corresponding base-pair/step parameters. This process is rigorous for atomic coordinates of base (and C1') atoms: running analyze on the rebuilt structure will yield the same set of parameters that users initially input. For more details, see the 2003 3DNA paper, the 2015 DSSR paper, and the DSSR User Manual.
The challenge lies in modeling the backbones. For right-handed A- or B-form DNA, users can build full-atomic models with canonical backbone conformations of C3'-endo or C2’-endo sugar conformations and anti glycosidic bonds. However, left-handed Z-DNA has unique structural features—such as syn-G, CpG, and GpC dinucleotides as building blocks instead of single nucleotides—that are not fully addressed by the 3DNA rebuild program.
DSSR (Pro version or the Academic v2.5.2) offers a solution by providing tools to build extended Z-DNA structures with proper backbones. The commands are as follows:
x3dna-dssr -i=1qbj.pdb1 --select-chains='D E' --delete-water -o=model.pdb
x3dna-dssr tasks -i=model.pdb --frame-pair=last -o=model1-ref-last.pdb
# poly d(GC) : poly d(GC)
x3dna-dssr fiber --z-dna --repeat=1 -o=conn.pdb
x3dna-dssr tasks -i=conn.pdb --frame-pair=first --remove-pair -o=ref-conn.pdb
x3dna-dssr tasks --merge-file='model1-ref-last.pdb ref-conn.pdb' -o=temp1.pdb
x3dna-dssr tasks -i=temp1.pdb --frame-pair=last --remove-pair -o=temp2.pdb
x3dna-dssr tasks -i=model.pdb --frame-pair=first -o=model1-ref-first.pdb
x3dna-dssr tasks --merge-file='temp2.pdb model1-ref-first.pdb' -o=duplicate-model.pdb
x3dna-dssr --order-residue -i=duplicate-model.pdb -o=temp3.pdb --po-bond=3.6
x3dna-dssr --renumber-residue -i=temp3.pdb -o=temp4.pdb
x3dna-dssr --connect-file -i=temp4.pdb -o=1qbj-duplicate.pdb --po-bond=3.6
The logic behind these commands is very straightforward, but technical details may look a bit complex for the uninitiated:
- The first command extracts the Z-DNA duplex consisting of chains D and E from PDB entry
1qbj.pdb1 (the first biological unit) and remove water molecules (model.pdb). The Z-DNA duplex has sequence: CGCGCG/CGCGCG.
- The next command sets the Z-DNA duplex (
model.pdb) into the reference frame of the last base pair, i.e., G-C (model1-ref-last.pdb).
- The
fiber model consists of the GpC dinucleotide step (conn.pdb), which is then set into the reference frame of the first base pair (G-C). The first G-C pair is removed from the coordinate file ref-conn.pdb which consists of only one C-G pair.
- The two PDB files,
model1-ref-last.pdb and ref-conn.pdb, share a common reference frame and are merged into a single PDB file (temp1.pdb).
- The merged PDB file (
temp1.pdb) is then set into the reference frame of last base pair(i.e., C-G) which is removed from the resulting coordinate file (temp2.pdb). Now the job of the GpC fiber connector is done.
- The Z-DNA duplex (
model.pdb) is once again set into the reference frame of the first base pair (i.e., C-G), leading to the coordinate file model1-ref-first.pdb.
- The two PDB files,
temp2.pdb and model1-ref-first.pdb, both consist of the same Z-DNA duplex but are in different orientations. They now share a common reference frame and are merged into the extended Z-DNA duplex (1qbj-duplicate.pdb).
- The last three commands (with options
--order-residue, --renumber-residue, --connect-file) are bookkeeping steps to ensure proper order and numbering of nucleotides along each chain, and generate the CONECT record for smooth view in PyMOL.
The final PDB coordinate file (1qbj-duplicate.pdb) can be downloaded, and visualized in DSSR-enabled cartoon-block representation as below:
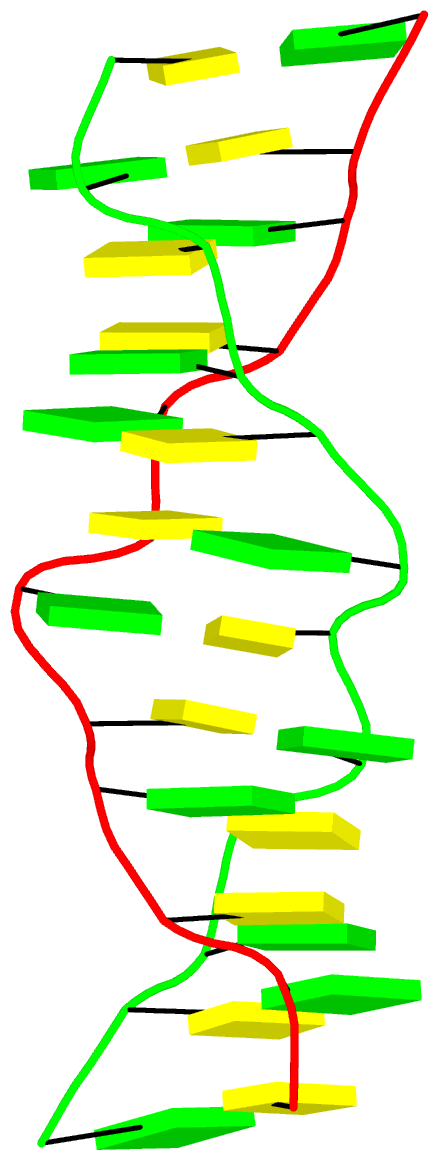
In January 29, 2025, I received the following email request from a long-time DSSR user:
... recently noted that 3DNA/DSSR automatically maps non-standard nucleotides to standard nucleotides. I wonder if you would be willing to share with us your most current version of mappings?
I responded to the user the same day, with detailed information about the mapping process in DSSR. The user was happy with my response, and that thread was quickly closed with a positive note.
On April 22, 2025, a related question, titled "Can x3dna-dssr correctly handle N1-methyl-pseudouridine?", was asked on the 3DNA Forum. In answering the question on the Forum, I referred to my email response to the previous user.
I now realize that writing a detailed blog post explaining the mapping process would be beneficial for DSSR users. It would also enable me to easily reference this blog post in future interactions with users.
3DNA/DSSR performs automatic mapping of modified nucleotides (including pseudouridine) to their standard counterparts. Over the years, the method has proven to work well in real-world applications. It is one of the defining features that make DSSR just work. For example, for the tRNA 1ehz, DSSR automatically identifies the following 14 modified nucleotides (of 11 unique types):
# x3dna-dssr -i=1ehz.pdb
List of 11 types of 14 modified nucleotides
nt count list
1 1MA-a 1 A.1MA58
2 2MG-g 1 A.2MG10
3 5MC-c 2 A.5MC40,A.5MC49
4 5MU-t 1 A.5MU54
5 7MG-g 1 A.7MG46
6 H2U-u 2 A.H2U16,A.H2U17
7 M2G-g 1 A.M2G26
8 OMC-c 1 A.OMC32
9 OMG-g 1 A.OMG34
10 PSU-P 2 A.PSU39,A.PSU55
11 YYG-g 1 A.YYG37
Users could run DSSR on a set of structures of interest, and collect the unique mappings for a complete list of modified nucleotides.
Moreover, DSSR has the --nt-mapping option that allows users to control the mapping process. The screenshot below is taken from the relevant part of the DSSR manual.
For example, DSSR automatically maps 5MU (5-methyluridine 5′-monophosphate) to t (i.e., modified thymine) because of the 5-methyl group. With the option --nt-mapping='5MU:u', DSSR would take 5MU as a modified uracil. This option allows for multiple mappings separated by comma. The mapping of 5MU to u or t is obviously arbitrary. DSSR is robust against the ambiguity in designating a modified nucleotide to its nearest canonical counterpart. For example, mapping 5MU to u or t has minimal influence on DSSR-derived base-pair parameters and other structural features.

Background information on the mapping
Over the years, I've refined the heuristics of the mapping process. In the early days with 3DNA, I kept an ever increasing list in file baselist.dat with hundreds of entries like: MIA a that maps MIA as a modified A, denoted as lowercase a. In recent releases of DSSR, I keep only the standard ones, with a total of 48 entries like ADE A, and DG5 G etc. If a residue is not a standard one, the following C function is called to do the mapping. DSSR performs filtering to decide if a residue is a nucleotide, and if so R (purine) or Y (pyrimidine).
static void derive_new_nt_std_name(long resi, struct_mol *pdb, char *info)
{
char str[BUF512];
double d1 = DMAX, d2 = DMAX;
long C1_prime, N1, C5;
struct_residue *r = &pdb->residues[resi];
if (r->type[RESIDUE_NT_UNKNOWN]) {
sprintf(r->std_name, "__%c", Gvars.abasic);
return;
}
if (is_R(resi, pdb)) { /* purine */
if (residue_has_atom(" O6 ", resi, pdb)) /* with ' O6 ' */
strcpy(r->std_name, "__g");
else if (!residue_has_atom(" N6 ", resi, pdb) && /* no ' N6 ' but ' N2 ' */
residue_has_atom(" N2 ", resi, pdb))
strcpy(r->std_name, "__g");
else
strcpy(r->std_name, "__a");
} else { /* a pyrimidine */
if (residue_has_atom(" N4 ", resi, pdb))
strcpy(r->std_name, "__c");
else if (residue_has_atom(" C7 ", resi, pdb))
strcpy(r->std_name, "__t");
else
strcpy(r->std_name, "__u");
C1_prime = find_atom_in_residue(" C1'", resi, pdb);
N1 = find_atom_in_residue(" N1 ", resi, pdb);
if (atoms_same_model_chain_altloc(C1_prime, N1, pdb))
d1 = dist_atoms(C1_prime, N1, pdb);
if (!dval_in_range(d1, 1.0, 2.0)) {
C5 = find_atom_in_residue(" C5 ", resi, pdb);
if (atoms_same_model_chain_altloc(C1_prime, C5, pdb))
d2 = dist_atoms(C1_prime, C5, pdb);
if (dval_in_range(d2, 1.0, 2.0))
strcpy(r->std_name, "__p");
}
}
if (!Gvars.standalone) {
sprintf(str, "\n\tmatched nucleotide '%s' to '%c' for %s\n"
"\tverify and add an entry in <baselist.dat>\n",
r->res_name, r->std_name[2], info);
logit(str);
}
}
Recently, I read carefully the two papers by Farag et al. on the ASC-G4 algorithm to calculate "advanced structural characteristics of G-quadruplexes" (2023), and the comprehensive analysis results of intramolecular G4 structures in the PDB (2024). By developing a convention to orient and number the four strands, ASC-G4 allows for unambiguous determination of the intramolecular G4 topology. It also has an in-depth discussion on assigning syn or anti glycosidic configuration of guanosines, and categorizes four different types of snapbacks.
I am glad to see that DSSR is cited in these two papers, as quoted below:
X3dna-DSSR (19) (http://x3dna.org) is a website that was created to calculate nucleic acid structural parameters, like the local base-pair parameters, local step base-pair parameters, torsion angles, etc, but not the special characteristics of G4. A subdomain dedicated to G4, DSSR-G4DB (Dissecting the Spatial Structure of RNA – G4 Data Base) (http://g4.x3dna.org) emanated from this website. It is a database that gathers and calculates some specific structural information about published G4s, like the topology, the rise, the helical twist, etc, but not the groove widths or the presence of snapbacks. -- Farag et al. (2023)
Indeed, DSSR-G4DB dose not classify snapbacks. I was aware of such non-canonical G4s when I first developed the G4 module in DSSR around 2017-2018, and the V-shaped loops was derived to reflect the peculiarity of snapbacks.
DSSR classifies groove widths as medium, wide, or narrow, based on the glycosidic angles of neighboring guanosines in a G-tetrad, following the G4 literature. Using PDB entry 2lod as an example, the relevant part of the DSSR output is shown below. The groove widths of the three G-tetrads in the G4-stem have the same pattern of groove=--wn, standing for medium, medium, wide, and narrow, respectively. Note that the medium groove is represented by a dash instead of m because --wn stands out more clearly than mmwn (similar idea applies to glycosidic bond, e.g., sss-).
1 glyco-bond=sss- sugar=---- groove=--wn Major-->WC N- nts=4 GGGG A.DG1,A.DG6,A.DG20,A.DG16
2 glyco-bond=---s sugar=---- groove=--wn WC-->Major N+ nts=4 GGGG A.DG2,A.DG7,A.DG21,A.DG15
3 glyco-bond=---s sugar=---- groove=--wn WC-->Major N+ nts=4 GGGG A.DG3,A.DG8,A.DG22,A.DG14
Since DSSR-G4DB is a database, the user cannot provide his own G4 structure, to obtain structural information. Hence the necessity of developing a website where the user uploads his G4 structure file to obtain all its important and specific structural characteristics (like the topology, the groove width, the tilt and twist angles, etc.). This can be very useful, not only for the analysis of published PDB structures but also for structures in refinement or obtained from MD simulations, to evaluate their quality. To our knowledge, there is no website dedicated to G4 to do such calculations in real-time. Therefore, we developed the algorithm ASC-G4 (advanced structural characteristics of G4) and deployed it as a user-friendly website at the following address: http://tiny.cc/ASC-G4. -- Farag et al. (2023)
Thanks to the NIH R24GM153869 grant support, the http://g4.x3dna.org website now allows users to upload their own atomic structures in PDB for mmCIF format for the identification, annotation, and visualization of G4s. See the example of uploading PDB coordinate file 2lod.pdb.
As background, I had long aspired to develop a dynamic website for on-demand G4 structural analysis but was unable to pursue this goal until recently. During the 4-year funding gap, I still managed to maintain the website g4.x3dna.org, which provides DSSR results for G4 structures in the PDB (a resource now known as the DSSR-G4DB database). To date, the only published work related to G4s is my 2020 paper on the integration of DSSR with PyMOL. Clearly, a dedicated method paper detailing the G4 module in DSSR and the g4.x3dna.org website has been long overdue.
As an initial step toward addressing this gap, I have recently revised the G4-related code in DSSR, fixed existing bugs, and added new features. The g4.x3dna.org website has undergone a complete overhaul, enabling users to upload their own structures for dynamic G4 analysis. Additionally, the DSSR-G4DB database is being actively updated on a weekly basis as new PDB entries are added.
Calculation of the twist and tilt angles. In G4, the helix twist is the rotation of a tetrad relative to its successive one. To measure the twist angle, the most spread method is that described by Lu and Olson (2003) (32) and Reshetnikov et al. (2010) (33). In this method, the angle is calculated from the dot product between two C1’–C1’ vectors from two successive tetrads, i and i + 1, the C1’ atoms of each vector belonging to two adjacent guanosines of a Hbp. The issue with this method is that it does not allow access to the sign of the angle, which defines the direction of the G4 helix, viz. right-handed or left-handed. -- Farag et al. (2023)
There is clearly a misunderstanding in the above text. 3DNA/DSSR can handle left-handed Z-DNA without any issues. DSSR also reports negative twist angles for left-handed G4s, as shown clearly for PDB entry 7d5e, for example.
3DNA/DSSR derives a complete of set of six base-pair parameters (including shear and opening), six step parameters (including twist and rise), and six helical parameters, using a rigorously defined and completely reversible algorithm (CEHS) and the standard base-reference frame. See section "3.2.3 Base pairs" in DSSR User Manual for more details. The DSSR output for G4s (as in DSSR-G4DB) reports only twist and rise, along with overlapped areas, simply because these are the most important parameters and easily interpretable.
The list of the resolved G4 structures was downloaded from the ONQUADRO website (https://onquadro.cs.put.poznan.pl/) (39) at about the end of October 2023. It consisted of 291 intramolecular structures (named unimolecular in the website) and 154 intermolecular G4s (96 bimolecular and 58 tetramolecular). Only the intramolecular structures were kept for this study. To this list, we added 55 missing intramolecular structures that were found on the website of DSSR-G4DB (http://g4.x3dna.org) (40). From the merged list, 345 structures were downloaded from the Protein Data Bank (PDB) (http://www.rscb.org/pdb/) (41) because one structure had no available coordinates in the PDB format (7ZJ5 (42)). -- Farag and Mouawad (2024)
DSSR adopts the frame of reference of Webba da Silva, designating the four strands and grooves of G4-stem as shown below using PDB entries 8ht7 (G1 in syc) and 5ua3 (G1 in anti) as example for the syn or anti glycosidic bond of the 5'-guanosine, respectively.
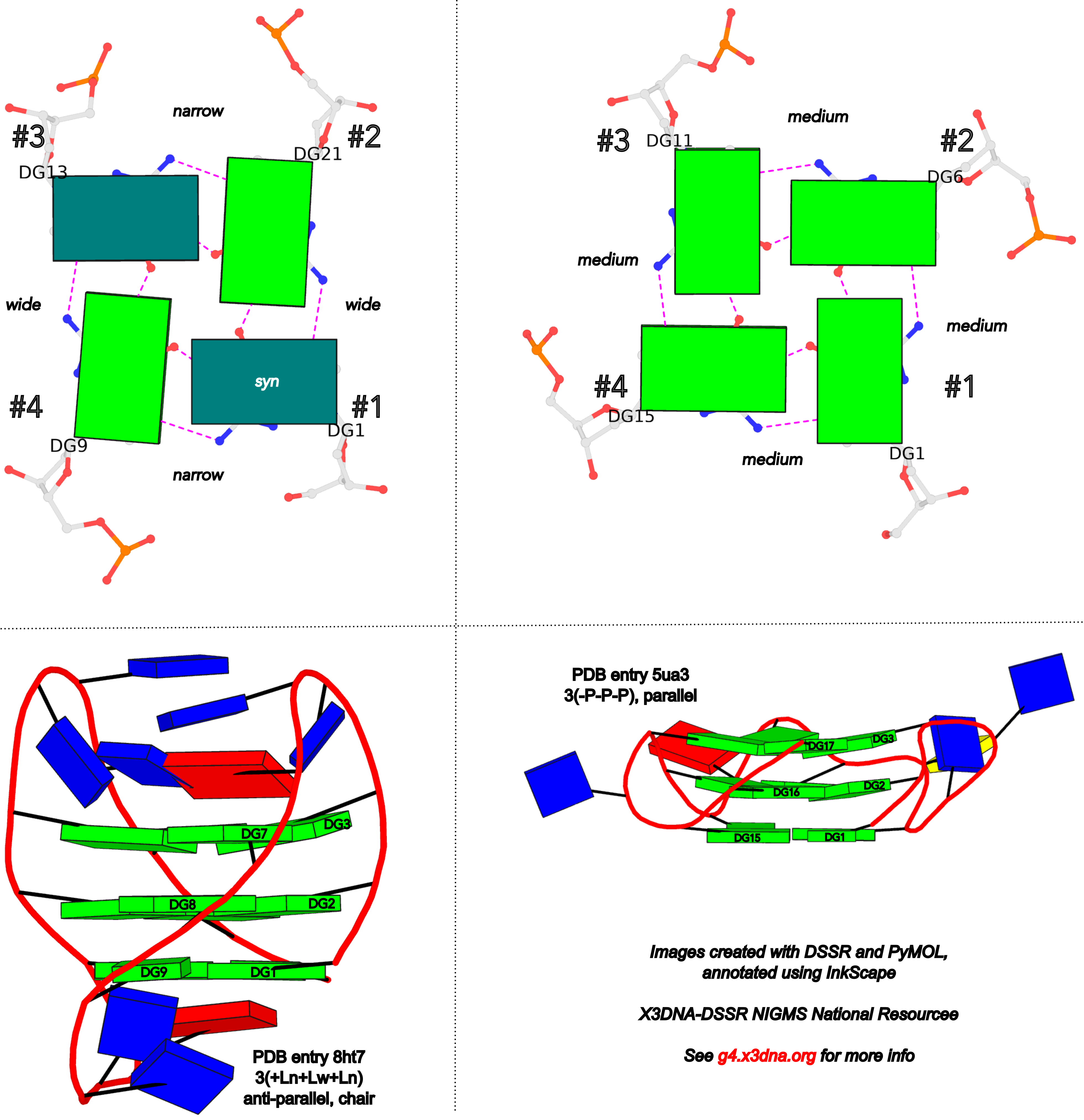
In DSSR, the first strand (#1) is always upward (U) from 5' to 3'-end, and the polarity of the other three strands is determined by its orientation relative to #1: U if parallel, or D if antiparallel. There are a total of 2x2x2=8 possible combinations of U and D for the three strands, which define parallel (U4: UUUU), antiparallel (U2D2: UDDU, UDUD, UUDD), or hybrid (UD3: UDDD; U3D: UDUU, UUDU, UUUD). For example, the PDB entry 2lod is characterized by DSSR as: "hybrid-(mixed), UUUD, U3D(3+1)", and PDB entry 8ht7 as: "anti-parallel, UDUD, chair(2+2)". This notation is topologically equivalent to the one adopted by ASC-G4 but with opposite orientation of the strands.
Overall, DSSR and ASC-G4 provide different perspectives on G4 structures. It is to the user to decide which one is more suitable for their needs.
References
Farag,M. et al. (2023) ASC-G4, an algorithm to calculate advanced structural characteristics of G-quadruplexes. Nucleic Acids Res., 51, 2087–2107.
Farag,M. and Mouawad,L. (2024) Comprehensive analysis of intramolecular G-quadruplex structures: furthering the understanding of their formalism. Nucleic Acids Res., gkae182.
In late September of 2018, I contacted Dr. Mateus Webba da Silva requesting a copy of his 2007 article, titled "Geometric formalism for DNA quadruplex folding". At that time, I had implemented a G4 module within DSSR for the automatic identification, comprehensive annotation, and schematic visualization of G-quadruplexes from 3D atomic coordinates. I noticed the 2007 paper, and was intrigued by the following sentences in the abstract:
A formalism is presented describing the interdependency of a set of structural descriptors as a geometric basis for folding of unimolecular quadruplex topologies. It represents a standard for interpretation of structural characteristics of quadruplexes, and is comprehensive in explicitly harmonizing the results of published literature with a unified language.
Mateus kindly sent me a copy of the 2007 article, and shortly afterwards he also shared with me the Dvorkin et al. (2018) paper on "Encoding canonical DNA quadruplex structure". I carefully read both papers, plus the Karsisiotis et al. (2013) tutorial paper. I was impressed by the elegance of the formalism: simple and systematic, so I immediately decided to add this feature to the G4 module of DSSR.
As the Chinese saying goes, "纸上得来终觉浅,绝知此事要躬行" ("What you learn from books is always shallow. You must practice it yourself to know it well." -- Google Translate). The implementation process was challenging because of subtleties in the formalism, but very rewarding. It is all about scientific understanding and software engineering. Only after a thorough understanding and attention to meticulous details can one create a robust and reliable software tool. On the other hand, once properly implemented, the DSSR G4 module can be applied consistently. Any discrepancies between DSSR output and literature merit further investigation. These discrepancies could either arise from bugs in DSSR (which I will promptly address upon identification) or, more likely, typos or errors in the reported results.
Webba da Silva (2007) systematically described the interdependency of glycosidic bond (syn or anti), strand polarity (parallel or anti-parallel), groove width (narrow, medium, or wide), and loop type (lateral, propeller, or diagonal) in unimolecular G-quadruplexes. Figures 1-3 and Scheme 1 of Webba da Silva (2007) are very informative, and easy to follow conceptually. The Karsisiotis et al. (2013) tutorial provided further details based on experimentally determined G-quadruplex structures from the PDB (e.g., Figure 3: the schematic for all possible combinations of glycosidic bond and the corresponding groove-width combinations in G-tetrad). Some key observations:
- Since glycosidic bond can be either syn or anti, there a total of
2x2x2x2 = 16 possible combinations in a G-tetrad.
- The disposition of glycosidic bond of guanosines in a G-tetrad leads to only eight possible groove-width combinations.
- Only tetrads with the same groove-width combinations may stack to form stable G-quadruplexes.
- Propeller loops invariably link medium grooves within a G-quadruplex stem.
- Lateral and diagonal loops bridge guanosines of different glycosidic bond.
- If a single-stranded quadruplex starts with a narrow groove, it can only be with a clockwise loop progression (i.e., +lateral).
- There are 26 permissible looping combinations within a canonical unimolecular G-quadruplex (G4-stem).
To unambiguously characterize a G4-stem, Webba da Silva (2007) defined a frame of reference where the 5’-G in a G4-stem is set as the origin, and the first strand is progressing towards the viewer. Regardless of the clockwise or anti-clockwise progression of the base sequence, the scheme designates one orientation for the syn and anti glycosidic bond by following G+G H-bonding alignments. Put another way, grooves and strands are strictly related to the reference (first) strand in an anti-clockwise manner, irrespective of the progression of the base sequence. The point is illustrated in the figure below, using PDB entries 8ht7 (G1 in syc) and 5ua3 (G1 in anti) as an example for the syn or anti glycosidic bond of the 5'-guanosine, respectively.
Based on previous work, the Dvorkin et al. (2018) paper proposed a systematic nomenclature for G4-stem. The single structural descriptor contains:
- The number of G-tetrads (i.e., the G-tract length).
- Loop types (lowercase l for lateral, p for propeller, and d for diagonal) and relative direction ("+" for clockwise and "-" for anti-clockwise progression, using the frame of reference described above).
- For lateral loops, the groove widths ("w" for wide, and “n” for narrow) are denoted in subscript.
So a complete descriptor could be 2(+lnd−p), as shown in Figure 1A of the Dvorkin et al. (2018) paper. Significantly, Figure 1B therein further gave structural descriptors for six experimentally determined G4-stems from the PDB. These examples, plus the ones in the supplementary materials, were used to validate my implementation of the systematic nomenclature in the G4 module of DSSR. My results agree with those in the Dvorkin et al. (2018) paper, except for two cases, which are discussed below.
- For PDB entry 2gku: 3(-p-ln-lw) (Dvorkin et al.) vs 3(-P-Lw-Ln) (DSSR), with swapped n (narrow) and w (wide) groove widths for both lateral loops.
- For PDB entry 2lod: 3(-pd+ln) (Dvorkin et al.) vs 3(-PD+Lw) (DSSR), with swapped n (narrow) and w (wide) groove width for the lateral loop.
Note that in DSSR, I am using uppercase L/P/D for lateral/propeller/diagonal loop types, and lowercase n/w for narrow/wide groove widths, respectively. Doing so distinguishes between the different loop types and groove widths in pure text format.
After careful examination of these discrepancies, I still couldn’t find any errors in my implementation. So I contacted Mateus for verification (in early October 2018). Thankfully, he quickly responded and acknowledged the mistakes for PDB entry 2gku in Dvorkin et al. (2018), saying "There can not be a –Ln after the –p." Clearly, the wrong descriptor for PDB entry 2gku in Dvorkin et al. (2018) was due to a typographical error. This example illustrates the power of a robust software tool like DSSR.
References
Dvorkin,S.A. et al. (2018) Encoding canonical DNA quadruplex structure. Sci. Adv., 4, eaat3007.
Karsisiotis,A.I. et al. (2013) DNA quadruplex folding formalism – A tutorial on quadruplex topologies. Methods, 64, 28–35.
Webba da Silva,M. (2007) Geometric formalism for DNA quadruplex folding. Chemistry A European J, 13, 9738–9745.
Over the past few years, the Szachniuk group has made several significant contributions to the field of structural bioinformatics of G-quadruplexes. The following five publications are particularly noteworthy, and I am glad to see that 3DNA/DSSR have been cited in all of them.
1. Zok et al. (2020) -- ElTetrado: a tool for identification and classification of tetrads and quadruplexes
The BMC Bioinformatics paper introduced the ElTetrado software tool for identifying and classifying G-tetrads in unimolecular G-quadruplex structures into ONZ taxonomy. Here DSSR is employed to identify base-pairs and base-stacking interactions.
ElTetrado processes PDB and mmCIF files to identify quadruplexes and their component tetrads in nucleic acid structures (Fig. 2). It applies DSSR [24] to collect the preliminary information about base pairs and stacking.
We recommend that, apart from ElTetrado, the users should download the DSSR binary [24] and place it in the same local directory. DSSR is utilized for the preliminary analysis of base pairs in the input 3D structure. Its local installation allows the users to control DSSR execution. For example, one can decide to pass --symmetry parameter to x3dna-dssr binary when dealing with X-ray structures, which is necessary for some quadruplexes.
As documented in the DSSR Manual, by default, DSSR reads in the first model of an NMR ensemble. A biological unit of X-ray crystal structures in the PDB may contain symmetry-related components formatted as a MODEL/ENDMDL delimited, NMR-like ensemble. In such cases, the --symmetry (or --symm) option is required for DSSR to process the entire biological unit.
For example, x3dna-dssr -i=4ms9.pdb1 --symm leads to the identification of 10 Watson-Crick base pairs in the biological unit of PDB entry 4ms9 (uploaded). The --symm option is now enabled for user-uploaded PDB files on the skmatic.x3dna.org website. Without the --symm option, DSSR would not find any Watson-Crick base pairs in 4ms9.pdb1 since MODEL#1 is single stranded.
Noticing the confusion users may have in using the --symm option, I have revised DSSR to check for overlapped residues. When all models in an NMR ensemble are taken as a whole with the --symm option, there will be overlapped residues. In such cases, DSSR will report a diagnostic message and proceed with the first model only. The final result is as if --symm has not been specified. Put another way, specifying --symm for an NMR ensemble does no harm to the analysis. For example, analyzing PDB entry 8xeq with the --symm option would have the following message and only the first model would be processed.
x3dna-dssr -i=8xeq.pdb --symm -o=8xeq.out
[i] You specified --symm, but the input file is an (NMR) ensemble
*** in the following, only the FIRST model will be processed ***
Alternatively, if the users do not want to have a local version of DSSR binary, they can obtain the DSSR output in JSON format from any place and use them as input data for ElTetrado (with --dssr-json parameter).
ElTetrado is started from the command line. The users enter the program name and either --pdb followed by an input file name (the file should be in PDB or mmCIF format), or --dssr-json followed by a path to JSON file generated by DSSR, or both switches at once.
The JSON output from DSSR can be obtained directly from the skmatic.x3dna.org website for pre-processed PDB entries or user-supplied coordinate files. For examples, for PDB entry 1ehz, the URL is http://skmatic.x3dna.org/pdb/1ehz/1ehz.json. Alternatively, users can use the web API to get the JSON file, as shown below:
# Pre-processed PDB entry:
curl http://skmatic.x3dna.org/api/pdb/1ehz/json
# With user-supplied PDB file
curl http://skmatic.x3dna.org/api -F 'model=@1ehz.pdb' -F 'type=json'
curl http://skmatic.x3dna.org/api -F 'url=https://files.rcsb.org/download/1ehz.pdb.gz' -F 'type=json'
2. Popenda et al. (2020) -- Topology-based classification of tetrads and quadruplex structures
This paper presents the ONZ scheme to clarify tetrads in unimolecular structures (see Figure 2 therein). Note that DSSR, with option --G4=ONZ, classifies G-tetrads into ONZ taxonomy. For example, the PDB entry 2gku has one G-tetrad (G3, G9, G17, G21) in the O- category, and two G-tetrads in O+.
Structures from both sets were analyzed using self-implemented programs along with DSSR software from the 3DNA suite (Lu et al., 2015). From DSSR, we acquired the information about base pairs and stacking.
3. Zurkowski et al. (2022) -- DrawTetrado to create layer diagrams of G4 structures
DrawTetrado generates static layer diagrams that represent structural data in a pseudo-3D perspective. The layer diagram is very informative and visually pleasing, and it complements the cartoon block schematics generated by the DSSR-PyMOL integration and the detailed DSSR characterization of G-quadruplexes.
So far, the only visual model designed for the 3D structure of quadruplexes is cartoon block schematics (Fig. 1C). These models are generated by DSSR-PyMOL integration and presented as static images of the structure viewed from six perspectives (Lu, 2020).
4. Adamczyk et al. (2023) -- WebTetrado: a webserver to explore quadruplexes in nucleic acid 3D structures
The topologies underlying the classification of quadruplexes and other parameters of their structures can be analyzed using a few computational tools. DSSR (7) was the first to target the detection of G-quadruplexes in 3D structure data saved in PDB and PDBx/mmCIF files and to describe their features. It runs systematically on all entries in the Protein Data Bank and collects motifs found in the DSSR-G4DB database. ElTetrado (8) can identify and analyze G4s and other kinds of tetrads and quadruplexes, classify them, and compute their parameters. It is the core of the computation pipeline running within the ONQUADRO database system (9). The most recent tool for processing atom coordinates in the search for quadruplexes is ASC-G4 (10). It calculates more features than DSSR and ElTetrado, but is limited to unimolecular quadruplexes and supports only the PDB format.
The example illustration in Figure 3 of the paper is on PDB entry 6h1k, the major G-quadruplex form of HIV-1 LTR (long terminal repeat). The layer diagram shown below, re-generated using the WebTetrado website, helps visualize the detailed characterization of DSSR very nicely.
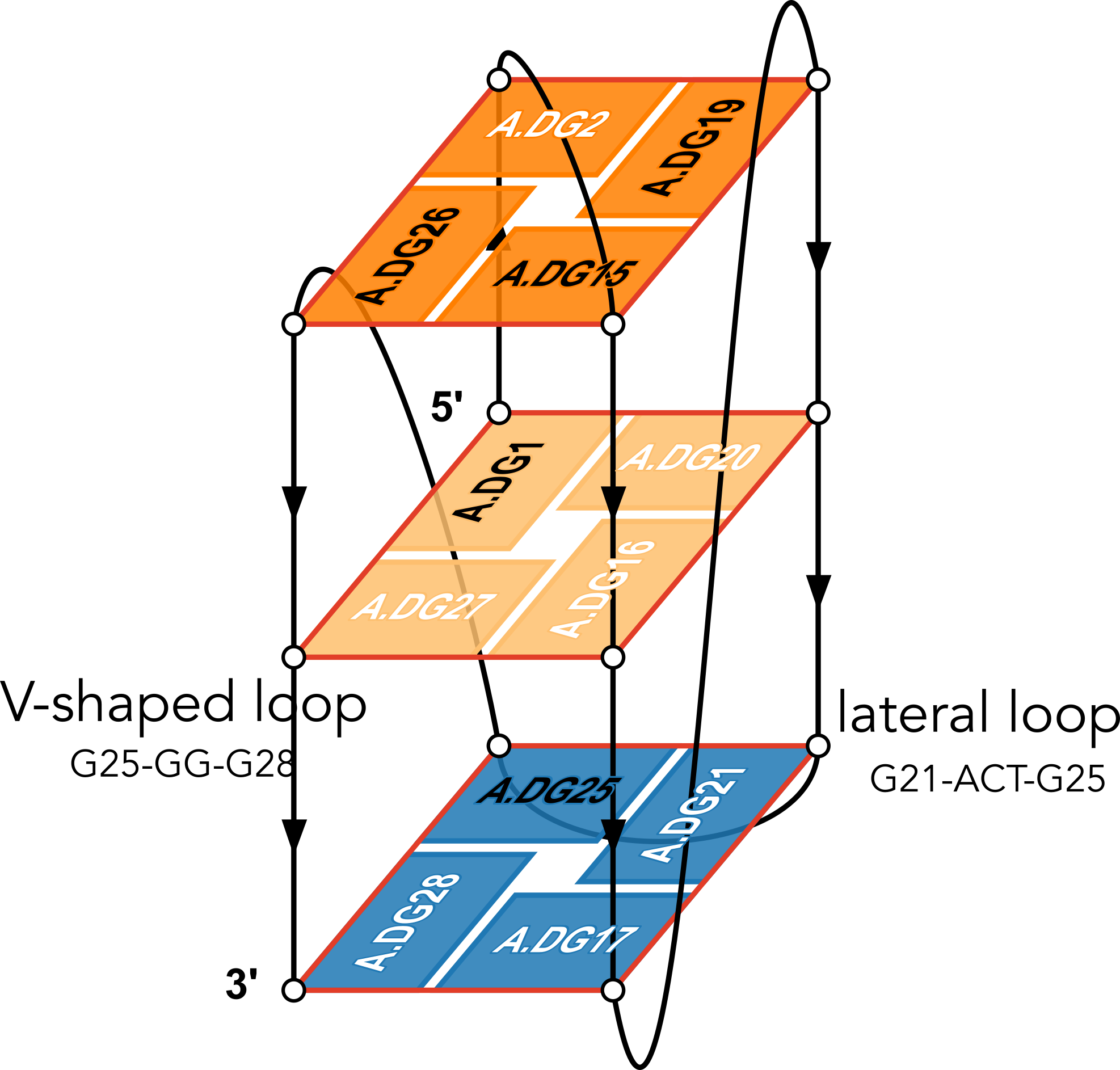 "In DSSR, a G4-helix is defined by stacking interactions of G-tetrads, regardless of backbone connectivity, and may contain more than one G4-stem." For PDB entry 6h1k, DSSR identifies a G-helix with three G-tetrads, ordered properly. Specifically, strand#1 consists of (G2, G1, and G25), even G1 and G25 are not covalently connected. On the other hand, "In DSSR, a G4-stem is defined as a G4-helix with backbone connectivity. Bulges are also allowed along each of the four strands." Thus, the G4-stem is composed of only two G-tetrads, as detailed below.
"In DSSR, a G4-helix is defined by stacking interactions of G-tetrads, regardless of backbone connectivity, and may contain more than one G4-stem." For PDB entry 6h1k, DSSR identifies a G-helix with three G-tetrads, ordered properly. Specifically, strand#1 consists of (G2, G1, and G25), even G1 and G25 are not covalently connected. On the other hand, "In DSSR, a G4-stem is defined as a G4-helix with backbone connectivity. Bulges are also allowed along each of the four strands." Thus, the G4-stem is composed of only two G-tetrads, as detailed below.
Stem#1, 2 G-tetrads, 3 loops, INTRA-molecular, UDDD, hybrid-(mixed), 2(D+PX), UD3(1+3)
1 glyco-bond=s--- sugar=---- groove=w--n Major-->WC Z- nts=4 GGGG A.DG1,A.DG20,A.DG16,A.DG27
2 glyco-bond=-sss sugar=.-.3 groove=w--n WC-->Major Z+ nts=4 GGGG A.DG2,A.DG19,A.DG15,A.DG26
step#1 mm(<>,outward) area=12.76 rise=3.47 twist=18.2
strand#1 U DNA glyco-bond=s- sugar=-. nts=2 GG A.DG1,A.DG2
strand#2 D DNA glyco-bond=-s sugar=-- nts=2 GG A.DG20,A.DG19
strand#3 D DNA glyco-bond=-s sugar=-. nts=2 GG A.DG16,A.DG15
strand#4 D DNA glyco-bond=-s sugar=-3 nts=2 GG A.DG27,A.DG26
loop#1 type=diagonal strands=[#1,#3] nts=12 GAGGCGTGGCCT A.DG3,A.DA4,A.DG5,A.DG6,A.DC7,A.DG8,A.DT9,A.DG10,A.DG11,A.DC12,A.DC13,A.DT14
loop#2 type=propeller strands=[#3,#2] nts=2 GC A.DG17,A.DC18
loop#3 type=diag-prop strands=[#2,#4] nts=5 GACTG A.DG21,A.DA22,A.DC23,A.DT24,A.DG25
List of 2 non-stem G4-loops (including the two closing Gs)
1 type=lateral helix=#1 nts=5 GACTG A.DG21,A.DA22,A.DC23,A.DT24,A.DG25
2 type=V-shaped helix=#1 nts=4 GGGG A.DG25,A.DG26,A.DG27,A.DG28
DSSR correctly identifies the 12-nt diagonal loop, containing a canonical duplex stem and a hairpin loop. Notably, the G-tetrad (25-21-17-28) does not belong to the G4-stem because of the broken backbone connectivity between G1 and G25. Instead, the Gs in the G-tetrad (25-21-17-28) become part of the following two loops, which are certainly unconventional yet follow naturally the DSSR definition of G4-stem.
- The propeller loop, which now includes G17 (part of the G-tetrad), in addition to C18.
- The unusual diag-prop (diagonal-propeller ) loop, which consists of G21, A22, C23, T24, and G25.
Moreover, DSSR also reports two loops that are not defined by the G4-stem: the V-shaped loop (G25-G26-G27-G28) and the lateral loop (G21-A22-C23-T24-G25). See the notes in the above layer diagram. V-shaped loop occurs when the 5’-endmost G-tetrad lies in the middle of the G-quartets stack as in the non-canonical G4 structures with snapbacks.
The G4-helix and G4-stem definitions parallel those for duplex helix and stem in DSSR. The characterization of loops follows naturally once G4-stem or duplex stem are identified. The unusual propeller and diagonal-propeller loops noted above are due to non-canonical structures, which also lead to the listing of non-stem G4-loops.
I may consider to add special handling of snapbacks (or other worthwhile classes of non-canonical G4 structures) so that the reported loops follow whatever consensus the community agrees upon in the future. Nevertheless, I would like to emphasize that the consistent definitions of G4-stem and loops in DSSR help pinpoint extraordinary features to draw users' attention to non-canonical G4 structures. The layer diagram from DrawTetrado and WebTetrado are very handy in illuminating the basic concept and technical details, as shown here for PDB entry 6h1k.
5. Zok et al. (2022) -- ONQUADRO: a database of experimentally determined quadruplex structures
The computational engine is composed of scripts utilising in-house and third-party procedures, responsible for data collection, quadruplex identification, computation of structure parameters, secondary structure annotation, visualisation of the secondary and tertiary structure models, database queries, generation of statistics, and newsletter preparation. DSSR (--pair-only mode) (36) and ElTetrado (39) functionalities are applied to identify quadruplexes, tetrads, and G4-helices in nucleic acid structures.
References
Adamczyk, B., Zurkowski, M., Szachniuk, M., & Zok, T. (2023). WebTetrado: a webserver to explore quadruplexes in nucleic acid 3D structures. Nucleic Acids Research, 51(W1), W607–W612. https://doi.org/10.1093/nar/gkad346
Popenda, M., Miskiewicz, J., Sarzynska, J., Zok, T., & Szachniuk, M. (2020). Topology-based classification of tetrads and quadruplex structures. Bioinformatics, 36(4), 1129–1134. https://doi.org/10.1093/bioinformatics/btz738
Zok, T., Kraszewska, N., Miskiewicz, J., Pielacinska, P., Zurkowski, M., & Szachniuk, M. (2022). ONQUADRO: a database of experimentally determined quadruplex structures. Nucleic Acids Research, 50(D1), D253–D258. https://doi.org/10.1093/nar/gkab1118
Zok, T., Popenda, M., & Szachniuk, M. (2020). ElTetrado: a tool for identification and classification of tetrads and quadruplexes. BMC Bioinformatics, 21(1), 40. https://doi.org/10.1186/s12859-020-3385-1
Zurkowski, M., Zok, T., & Szachniuk, M. (2022). DrawTetrado to create layer diagrams of G4 structures. Bioinformatics, 38(15), 3835–3836. https://doi.org/10.1093/bioinformatics/btac394