Announcing wDSSR: The Next-Generation Web Interface to X3DNA-DSSR
Dear 3DNA/DSSR Community,
We are thrilled to announce the official launch of wDSSR (https://web.x3dna-dssr.org/), the powerful new web interface to the X3DNA-DSSR analytical engine.
Developed by Drs. Shuxiang Li and Xiang-Jun Lu and supported by NIH grant R24GM153869, wDSSR represents a major leap forward from our highly popular 2019 Web 3DNA 2.0 framework. While Web 3DNA 2.0 has faithfully served the community for the analysis, visualization, and modeling of 3D nucleic acid structures, wDSSR was built from the ground up to take full advantage of modern web technologies and the latest DSSR backend capabilities.
A Modern, Streamlined Scientific Workflow
We have completely overhauled the user interface to provide a clean, intuitive, and task-driven experience. The core modeling and analysis tools are now seamlessly organized into a logical, single-word scientific workflow: Analyze, Rebuild, Model, Circularize, Mutate, Assemble, and Visualize.
Spotlight Feature: The "Assemble" Module
One of the most exciting upgrades is the newly renamed Assemble tab (formerly "Composite"). This advanced composite model builder allows you to effortlessly construct complex, higher-order models by linking any combination of nucleic acid duplexes or protein-DNA/RNA complexes. You can quickly connect up to six distinct target structures, ranging from simple linked A-DNA and B-DNA duplexes to large, protein-decorated structural assemblies.
Immediate Global Adoption
Although wDSSR has just launched, we are incredibly humbled to share that it is already seeing rapid worldwide adoption! According to recent network infrastructure data, the new interface is actively being used by researchers across North America, South America, Europe, and Asia. Within just a few days, we have recorded active sessions from prestigious institutions around the globe, including:
- The Weizmann Institute of Science in Israel
- Katholieke Universiteit Leuven in Belgium
- Queen's University in Canada
- Universidad Nacional Autonoma de Mexico (UNAM) in Mexico
- Emory University and the Wadsworth Centers Laboratories and Research in the United States
- Jawaharlal Nehru University and the China Education and Research Network in Asia
How to Cite
While a dedicated paper for wDSSR is currently in preparation, researchers should cite the server using its URL (https://web.x3dna-dssr.org/) alongside the 2019 Web 3DNA 2.0 paper and the foundational 2015 DSSR paper. Full details and funding acknowledgements can be found on our newly consolidated About page.
We invite you all to try out the new wDSSR platform! As always, your feedback is invaluable to us, and we encourage you to share your thoughts, questions, and structural models via the newly updated Questions & Feedback link in the wDSSR footer.
Happy modeling!
Among the rich set of RNA structural features derived by DSSR, the section of “List of stacks” apparently has not drawn much attention from the user community. As noted in the DSSR output,
a stack is an ordered list of nucleotides assembled together via base-stacking interactions, regardless of backbone connectivity. Stacking interactions within a stem are not included.
As always, the concept is best illustrated via concrete examples. Shown below are two such base stacks automatically identified by DSSR in the PDB entry 4p5j, the crystal structure of the tRNA-mimic from Turnip Yellow Mosaic Virus (TYMV) which was analyzed in detail in the 2015 DSSR NAR paper
 |
 |
| This critical linchpin in the tRNA mimic is stabilized by extensive base-stacking interactions. |
The intricate interactions between the D- and T-loops in the tRNA mimic include a five-base stack. |
The DSSR-introduced schematic block representation makes the base-stacking interactions immediately obvious. One can even easily discern the identity of bases, given the color-coding convention: A-red; C-yellow; G-green; T-blue; U-cyan. For example, the five stacked bases involved in the interaction of the D- and T-loops are: CAAAC
Moreover, longer and more complicate base-stacks can also be auto-detected by DSSR, as shown below for the asymmetric unit of PDB entry 1j8g, the crystal structure of an RNA quadruplex r(UGGGGU)4 at 0.61 Å resolution. Here DSSR identifies two 10-base stacks, each of UGGGGGGGGU (UG8U).

The corresponding DSSR output is as below:
List of 2 stacks
Note: a stack is an ordered list of nucleotides assembled together via
base-stacking interactions, regardless of backbone connectivity.
Stacking interactions within a stem are *not* included.
1 nts=10 UGGGGGGGGU A.U6,A.G5,A.G4,A.G3,A.G2,C.G22,C.G23,C.G24,C.G25,C.U26
2 nts=10 UGGGGGGGGU B.U16,B.G15,B.G14,B.G13,B.G12,D.G32,D.G33,D.G34,D.G35,D.U36

G-quadruplexes (hereafter referred to as G4) are a common type of higher-order DNA and RNA structures formed from G-rich sequences. The building block of G4 is a tetrad of guanines in a cyclic planar alignment, with four G+G pairs (cW+M type, see Figure below). A G4 structure is formed by stacking of G-tetrads and stabilized by cations at the center of the layers. G4 structures are polymorphic: the four strands can be parallel or anti-parallel, and loops connecting them can be of different types: lateral (edgewise), diagonal, or propeller (double-chain reversal). Moreover, G4 structures can be intra- or intermolecular, and even contain bulges.
From its initial releases, DSSR was able to detect G-tetrads, and listed them in a separate section. As of v1.7.0-2017oct19, DSSR has integrated existing features and created a new module to automatically identify and fully characterize G4 structures. The underlying algorithms have been further refined in v1.7.1-2017nov01, which was tested against all nucleic-acid-containing structures in the PDB.
Characterizations of three representative G4 examples (PDB entries 2m4p, 2hy9, and 5hix) are shown below, illustrating salient features (e.g., different types of loops) automatically extracted by DSSR.
2m9p
stem#1[#1] layers=3 INTRA-molecular parallel bulged-strands=1
1 syn=---- WC-->Major area=8.38 rise=3.64 twist=33.34 nts=4 GGGG A.DG3,A.DG8,A.DG12,A.DG16
2 syn=---- WC-->Major area=10.73 rise=3.23 twist=32.42 nts=4 GGGG A.DG5,A.DG9,A.DG13,A.DG17
3 syn=---- WC-->Major nts=4 GGGG A.DG6,A.DG10,A.DG14,A.DG18
strand#1* +1 DNA syn=--- nts=3 GGG A.DG3,A.DG5,A.DG6 bulged-nts=1 T A.DT4
strand#2 +1 DNA syn=--- nts=3 GGG A.DG8,A.DG9,A.DG10
strand#3 +1 DNA syn=--- nts=3 GGG A.DG12,A.DG13,A.DG14
strand#4 +1 DNA syn=--- nts=3 GGG A.DG16,A.DG17,A.DG18
loop#1 type=propeller strands=[#1,#2] nts=1 T A.DT7
loop#2 type=propeller strands=[#2,#3] nts=1 T A.DT11
loop#3 type=propeller strands=[#3,#4] nts=1 T A.DT15
2hy9
stem#1[#1] layers=3 INTRA-molecular anti-parallel
1 syn=ss-s Major-->WC area=13.69 rise=3.14 twist=19.08 nts=4 GGGG 1.DG4,1.DG10,1.DG18,1.DG22
2 syn=--s- WC-->Major area=13.40 rise=3.05 twist=28.05 nts=4 GGGG 1.DG5,1.DG11,1.DG17,1.DG23
3 syn=--s- WC-->Major nts=4 GGGG 1.DG6,1.DG12,1.DG16,1.DG24
strand#1 +1 DNA syn=s-- nts=3 GGG 1.DG4,1.DG5,1.DG6
strand#2 +1 DNA syn=s-- nts=3 GGG 1.DG10,1.DG11,1.DG12
strand#3 -1 DNA syn=-ss nts=3 GGG 1.DG18,1.DG17,1.DG16
strand#4 +1 DNA syn=s-- nts=3 GGG 1.DG22,1.DG23,1.DG24
loop#1 type=propeller strands=[#1,#2] nts=3 TTA 1.DT7,1.DT8,1.DA9
loop#2 type=lateral strands=[#2,#3] nts=3 TTA 1.DT13,1.DT14,1.DA15
loop#3 type=lateral strands=[#3,#4] nts=3 TTA 1.DT19,1.DT20,1.DA21
5hix
stem#1[#1] layers=4 inter-molecular anti-parallel
1 syn=s--s Major-->WC area=12.93 rise=3.64 twist=16.82 nts=4 GGGG A.DG1,B.DG4,A.DG12,B.DG9
2 syn=-ss- WC-->Major area=18.96 rise=3.71 twist=35.87 nts=4 GGGG A.DG2,B.DG3,A.DG11,B.DG10
3 syn=s--s Major-->WC area=15.16 rise=3.64 twist=18.64 nts=4 GGGG A.DG3,B.DG2,A.DG10,B.DG11
4 syn=-ss- WC-->Major nts=4 GGGG A.DG4,B.DG1,A.DG9,B.DG12
strand#1 +1 DNA syn=s-s- nts=4 GGGG A.DG1,A.DG2,A.DG3,A.DG4
strand#2 -1 DNA syn=-s-s nts=4 GGGG B.DG4,B.DG3,B.DG2,B.DG1
strand#3 -1 DNA syn=-s-s nts=4 GGGG A.DG12,A.DG11,A.DG10,A.DG9
strand#4 +1 DNA syn=s-s- nts=4 GGGG B.DG9,B.DG10,B.DG11,B.DG12
loop#1 type=diagonal strands=[#1,#3] nts=4 TTTT A.DT5,A.DT6,A.DT7,A.DT8
loop#2 type=diagonal strands=[#2,#4] nts=4 TTTT B.DT5,B.DT6,B.DT7,B.DT8

The molecular structure of the G-tetrad and two G4 structures in schematics representation. Upper left: atomic structure of G-tetrad, the building block of G4 structures. Here the green ‘square’ is created by connecting the C1’ atoms of the guanosines, and it is used to simplify the representation of G4 structures of PDB entries 2m4p (lower left) and 5dww (right). Note that the asymmetric unit of 5dww contains four biological units, which are coaxially stacked in two columns.
The DSSR output for PDB entry 5dww is listed below, showing the differences of a G4-helix vs. a G4-stem.
5dww
Note: a G4-helix is defined by stacking interactions of G4-tetrads, regardless
of backbone connectivity, and may contain more than one G4-stem.
helix#1[#2] layers=6 inter-molecular stems=[#1,#2]
1 syn=---- WC-->Major area=10.64 rise=3.54 twist=28.10 nts=4 GGGG A.DG3,A.DG7,A.DG11,A.DG16
2 syn=.--- WC-->Major area=11.63 rise=3.65 twist=31.14 nts=4 GGGG A.DG2,A.DG6,A.DG10,A.DG15
3 syn=---- WC-->Major area=28.36 rise=3.31 twist=-9.78 nts=4 GGGG A.DG1,A.DG5,A.DG9,A.DG14
4 syn=---- Major-->WC area=11.60 rise=3.75 twist=29.43 nts=4 GGGG C.DG1,C.DG14,C.DG9,C.DG5
5 syn=---- Major-->WC area=10.35 rise=3.49 twist=28.74 nts=4 GGGG C.DG2,C.DG15,C.DG10,C.DG6
6 syn=---- Major-->WC nts=4 GGGG C.DG3,C.DG16,C.DG11,C.DG7
strand#1 DNA syn=-.---- nts=6 GGGGGG A.DG3,A.DG2,A.DG1,C.DG1,C.DG2,C.DG3
strand#2 DNA syn=------ nts=6 GGGGGG A.DG7,A.DG6,A.DG5,C.DG14,C.DG15,C.DG16
strand#3 DNA syn=------ nts=6 GGGGGG A.DG11,A.DG10,A.DG9,C.DG9,C.DG10,C.DG11
strand#4 DNA syn=------ nts=6 GGGGGG A.DG16,A.DG15,A.DG14,C.DG5,C.DG6,C.DG7
......
List of 4 G4-stems
Note: a G4-stem is defined as a G4-helix with backbone connectivity.
Bulges are also allowed along each of the four strands.
stem#1[#1] layers=3 INTRA-molecular parallel
1 syn=---- WC-->Major area=11.63 rise=3.65 twist=31.14 nts=4 GGGG A.DG1,A.DG5,A.DG9,A.DG14
2 syn=.--- WC-->Major area=10.64 rise=3.54 twist=28.10 nts=4 GGGG A.DG2,A.DG6,A.DG10,A.DG15
3 syn=---- WC-->Major nts=4 GGGG A.DG3,A.DG7,A.DG11,A.DG16
strand#1 +1 DNA syn=-.- nts=3 GGG A.DG1,A.DG2,A.DG3
strand#2 +1 DNA syn=--- nts=3 GGG A.DG5,A.DG6,A.DG7
strand#3 +1 DNA syn=--- nts=3 GGG A.DG9,A.DG10,A.DG11
strand#4 +1 DNA syn=--- nts=3 GGG A.DG14,A.DG15,A.DG16
loop#1 type=propeller strands=[#1,#2] nts=1 T A.DT4
loop#2 type=propeller strands=[#2,#3] nts=1 T A.DT8
loop#3 type=propeller strands=[#3,#4] nts=2 TT A.DT12,A.DT13
--------------------------------------------------------------------------
stem#2[#1] layers=3 INTRA-molecular parallel
1 syn=---- WC-->Major area=11.60 rise=3.75 twist=29.43 nts=4 GGGG C.DG1,C.DG5,C.DG9,C.DG14
2 syn=---- WC-->Major area=10.35 rise=3.49 twist=28.74 nts=4 GGGG C.DG2,C.DG6,C.DG10,C.DG15
3 syn=---- WC-->Major nts=4 GGGG C.DG3,C.DG7,C.DG11,C.DG16
strand#1 +1 DNA syn=--- nts=3 GGG C.DG1,C.DG2,C.DG3
strand#2 +1 DNA syn=--- nts=3 GGG C.DG5,C.DG6,C.DG7
strand#3 +1 DNA syn=--- nts=3 GGG C.DG9,C.DG10,C.DG11
strand#4 +1 DNA syn=--- nts=3 GGG C.DG14,C.DG15,C.DG16
loop#1 type=propeller strands=[#1,#2] nts=1 T C.DT4
loop#2 type=propeller strands=[#2,#3] nts=1 T C.DT8
loop#3 type=propeller strands=[#3,#4] nts=2 TT C.DT12,C.DT13
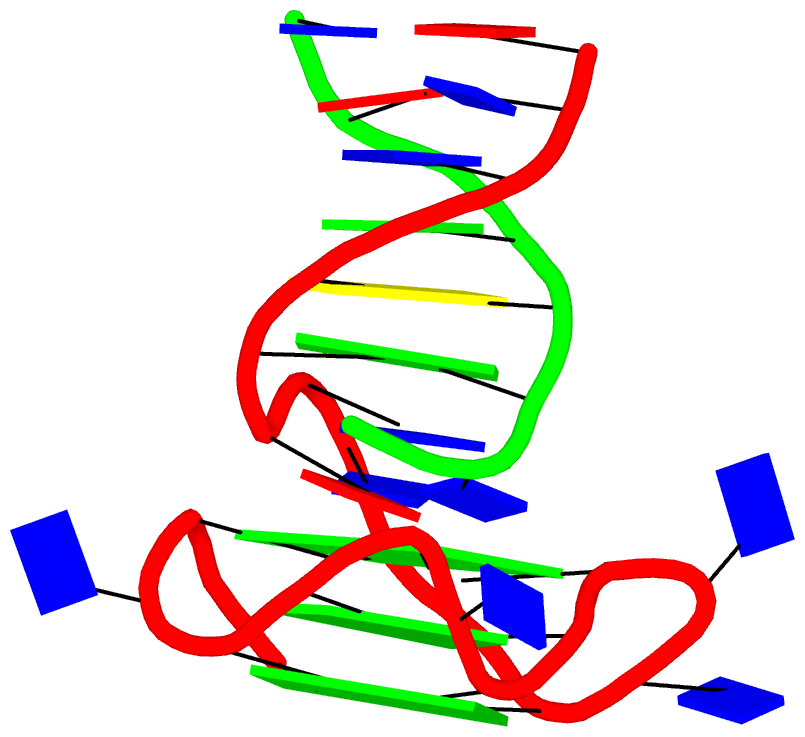
In addition to base pairs, DSSR also automatically detects higher-order base associations. They are generally termed multiplets, consisting of three or more co-planar bases arranged together via H-bonding interactions. The simplest multiplets are base triplets. For example, the yeast phenylalanine tRNA (PDB entry 1ehz) contains four base triplets, as shown below:

The well-known (types I and II) A-minor motifs are also multiplets of three bases. Similarly, the G-tetrad where four guanine bases associate via Hoogsteen H-bonding to form a square planar structure is also a special multiplet. The G-tetrad is the building block of the G-quadruplexes. As of v1.7.0-2017oct19, DSSR can automatically identify and characterize G-quadruplexes (see the DSSR User Manual).
The DSSR algorithm for detecting multiplets is generally applicable. It can identify as many co-planar bases as available in a given structure. Shown below is an octad, consisting of a G-tetrad in the middle and four Us on the peripheries. The octad is derived from PDB entry 1j8g using atomic coordinates from biological assembly 1 and 3.

The DSSR-Jmol paper, titled "DSSR-enhanced visualization of nucleic acid structures in Jmol", has been officially published in the 2017 web-server issue of Nucleic Acids Research (NAR). Notably, the work has been featured in the cover image, as shown below:

Caption: 3D interactive visualization of selected RNA structural features enabled by the DSSR-Jmol integration (http://jmol.x3dna.org). Clockwise from upper left: Structure of the xpt-pbuX guanine riboswitch in complex with hypoxanthine (PDB id: 4fe5) in ‘base blocks’ representation. The three-way junction loop encompassing the metabolite (in space-filling representation) is color-coded by base identity: A, red; C, yellow; G, green; U, cyan. The loop-loop interaction (a kissing-loop motif) at the top is highlighted in red (upper left corner). Structure of the Thermus thermophilus 30S ribosomal subunit in complex with antibiotics (PDB id: 1fjg) in step diagram. The 16S ribosomal RNA is color-coded in spectrum with the 5′-end in blue and the 3′-end in red (upper middle). Structure of the classic L-shaped yeast phenylalanine tRNA (PDB id: 1ehz) in step diagram, with the three hairpin loops highlighted in red and the [2,1,5,0] four-way junction loop in blue (upper right corner). Structure of the Pistol self-cleaving ribozyme (PDB id: 5ktj), showcasing (in red) the horizontal helix in space-filling representation. The helix is composed of six short stems stabilized via coaxial stacking interactions (bottom).
The DSSR-Jmol integration bridges the DSSR command-line analyzing tool and the Jmol molecular viewer seamlessly together via the standard JSON interface. Now users can select DSSR-derived RNA structural features (such as base pairs, double helices, various loops, etc.) and visualize them in novel representations in Jmol interactively. Moreover, fine-grained characteristics of these features can be queried via the Jmol SQL for DSSR. The DSSR-Jmol integration fills a gap in RNA structural bioinformatics, and brings RNA visualization to an entirely new level. The web interface (http://jmol.x3dna.org) is fully functional and easy to use, serving a huge user base of researchers, educators, and students alike.
Featured as the cover image of the 2017 NAR web-server issue, DSSR's publicity would surely increase through the DSSR-Jmol integration. Additionally, I've written a new post (on the 3DNA Forum) that provides the scripts and datafiles used to create the cover image.
I am pleased to announce the (advance online, May 3, 2017) publication of a new paper titled "DSSR-enhanced visualization of nucleic acid structures in Jmol" in Nucleic Acids Research (NAR). Co-authored by Robert Hanson (Jmol) and me (DSSR), the article will appear in the July 2017 web-server issue of NAR. Here are the key links related to the paper:
The DSSR-Jmol integration project was initiated in October 2013 when I approached Bob at a meeting organized by RCSB PDB at Rutgers. Thereafter, we met only once in July 2014 in Paris. Over the years, we have mostly communicated via email, occasionally facilitated by Skype. Our work bridges the DSSR command-line analyzing tool and the Jmol molecular viewer together via a simple JSON interface and a powerful query language. Users can now select DSSR-derived RNA structural features (such as base pairs, double helices, and various loops) as easily as they can select protein alpha-helices and beta-strands. Moreover, fine-grained characteristics of these features can be queried via Jmol SQL for DSSR (see examples below). Notably, the novel representation styles (step diagram and base blocks) and coloring schemes bring RNA visualization to an entirely new level (see Figure 3 of the paper).
load =1ehz/dssr # load yeast phenylalanine tRNA to Jmol with DSSR annotation
SELECT hairpins # select the three hairpin loops
SELECT junctions # select the four-way junction loop
select within(dssr, "nts WHERE is_modified") # select modified nucleotides (14 total)
SELECT within(dssr, "pairs WHERE name != 'WC'") # select non-Watson-Crick pairs
SELECT within(dssr, "pairs WHERE name = 'WC' OR name = 'Wobble'") # select canonical pairs
Select within(dssr, "pairs WHERE name != 'WC' AND name != 'Wobble'") # select non-canonical pairs
SELECT within(dssr, "pairs WHERE LW = 'tSW'") # select pairs of type tSW per Leontis-Westhof
The DSSR-Jmol integration fills a gap in RNA structural bioinformatics, serving a huge user base of researchers, educators, and students alike. Its functionality is freely accessible either via the Jmol application, or the JSmol-based website (http://jmol.x3dna.org). By adhering to web standards, the website is fully functional in all modern browsers on various computer/operating systems (including handheld devices, such as tablets and smart phones). The web interface is simple and intuitive, and new users can get started easily. It also allows power users to take full advantage of Jmol scripting via a command-line console.
This work also provides an example for integrating DSSR-derived features into other molecular graphics programs or bioinformatics pipelines involving nucleic acid structures. By design, DSSR is a stand-alone, command-line program written in ANSI C. The binary executables are only ~1MB in size, and self-contained. With zero dependencies, no setup or configuration, it is trivial to get DSSR up and running. DSSR uncovers a wide range of RNA/DNA structural features in a consistent, easily accessible framework. It possesses a much richer set of functionalities for nucleic acid structural analysis (see the DSSR User Manual) than any other existing tools I am aware of. Moreover, the program is efficient and robust, making it an ideal component to be integrated into other pipelines, especially via the standard and structured JSON interface.
Collaborating with Bob has been a truly exciting experience. The NAR-web publication represents a gratifying intermediate result along an on-going journey. Hopefully, others (may be some of you) can join us in pushing forward the field of RNA structural bioinformatics.
Recently, while analyzing a representative set of RNA structures from the PDB, I came across three weird entries. They are documented below, primarily for my own record.
- 5els — “Structure of the KH domain of T-STAR in complex with AAAUAA RNA”. There are two alternative conformations for the six-nt
AAAUAA RNA component, labeled A and B, respectively. Normally, the A/B alternative coordinates for each atom are put directly next to each other, and assigned the same chain id, as in 1msy for the phosphate group of G2669 on chain A. In 5els, however, the two alternative conformations (A/B) are separated into two chains: chain H for A, and chain I for B.
- 1vql — “The structure of the transition state analogue ‘DCSN’ bound to the large ribosomal subunit of Haloarcula marismortui”. The three-nt fragment DA179—C180—C181 on chain 4 is in the 3’—>5’ direction.
- 4r3i — “The crystal structure of m(6)A RNA with the YTHDC1 YTH domain”. The mmCIF file has a model number of 0, instead of 1 (as in other cases I am aware of).
Dear 3DNA Forum subscribers,
Here are some highlights of recent developments of 3DNA/DSSR:
Note: If you’ve difficulty in accessing the 3DNA homepage, possibly the case from mainland China (as I know it), please visit its duplicate at http://home.x3dna.org. This newsletter is written in Markdown, with a translated HTML version posted on the 3DNA homepage.
3DNA v2.3
The C source code is now available. Since the programs are written in strict ANSI C, 3DNA can be compiled (as is) on any computers/operating systems with a C (or C++) compiler. For user convenience, three binary distributions (with source code under the src/ subdirectory) are provided for Windows, Linux, and Mac OS X. The distributed Windows version works in native Windows (7 and up, via the cmd command-line interface, or ConEMU), MinGW/Msys (Msys2), and Cygwin, in either 32 or 64-bit.
A new set of ‘simple’ base-pair and step parameters was introduced to give ‘intuitive’ numerical values for non-Watson-Crick base pairs and associated steps. See the short communication titled Characterization of base pair geometry in the January 2016 issue of Computational Crystallography Newsletter (CCN).
The fiber program includes a new option, --pauling, for easy generation of Pauling & Corey triplex models of DNA/RNA with arbitrary base sequence. See my blogpost titled Pauling’s triplex model of nucleic acids is available in 3DNA.
Thomas Holder (PyMOL Principal Developer at Schrödinger, Inc.) has built a PyMOL wrapper to 3DNA fiber models. Now generating standard, regular DNA/RNA models in PyMOL is straightforward — thanks, Thomas!
DSSR (Dissecting the Spatial Structure of RNA)
Selected features of DSSR have been incorporated into Jmol (in collaboration with Robert Hanson, Jmol Principal Developer), and PyMOL (in collaboration with Thomas Holder). In Jmol application (via the Console window), one can now, for example, load =1ehz/dssr and then select hairpins; color red to see where the three hairpin loops are in 3D. The Jmol-DSSR web interface makes DSSR-enhanced visualization of nucleic acid structures in Jmol readily accessible to a broad user base, and has been employed in classes for educational purpose. A sample image of DSSR-derived cartoon-block representation via PyMOL is available for PDB entry 5dww, which has a G-quadruplex-duplex interface.
Since the publication of the Nucleic Acids Research paper in 2015, DSSR has been continuously refined and expanded, with a total of 36 new releases (from v1.2.8 to v1.6.4) as of this writing. Notably, the --json option provides DSSR-derived parameters in the simple, structured, and standard JSON format that can be easily parsed. This JSON output format is the (preferred) way for the outside world to interface with DSSR, and the Jmol-DSSR integration is built upon it. The --nmr option allows for batch processing of MODEL/ENDMDL-delineated NMR ensembles or trajectories of molecular dynamics (MD) simulations. Did you know that scripts and data files for reproducing the reported results are available in the DSSR-NAR paper section on the 3DNA Forum?
The User Manual is now 88-page long, covering nevertheless only the most common use cases of what DSSR has to offer. Miss a feature that you would like to have? Maybe it is already there or can be easily implemented in DSSR. Simply ask (on the 3DNA Forum), and I’ll try my best to help.
SNAP (Structures of Nucleic Acid-Protein complexes)
- SNAP aims to consolidate, refine, and significantly extend commonly used functionalities for DNA/RNA-protein structural analysis in one easy-to-use program. Currently in beta testing, SNAP is already fully functional, with features for characterizing the protein-nucleic acid interface and identifying amino acid-base pairing and stacking interactions.
A note for 3DNA/DSSR users in mainland China: It’s a pleasure to see the ~100 registrations on the 3DNA Forum with emails ending in .cn, 163.com, or qq.com etc., mostly from recent years. I’m planning a trip to China in 2017, and I’d be happy to meet some of you for academic exchanges and possible collaborations (学术交流、合作). If you’re interested, let’s get in touch!
Best regards,
Xiang-Jun
—
Dr. Xiang-Jun Lu (律祥俊)
Email: xiangjun@x3dna.org
Web: http://x3dna.org/
Forum: http://forum.x3dna.org/
In 1953, Pauling and Corey published an influential paper, titled A proposed structure for the nucleic acids, in Proc. Natl. Acad. Sci. (PNAS). Key features of the proposed model is summarized in their Letter to Nature, Structure of the Nucleic Acids, published in Nature on February 21, 1953.
We have formulated a structure for the nucleic acids which is compatible with the main features of the X-ray diagram and with the general principles of molecular structure, and which accounts satisfactorily for some of the chemical properties of the substances. The structure involves three intertwined helical polynucleotide chains. Each chain, which is formed by phosphate di-ester groups and linking β-D-ribofuranose or β-D-deoxyribofuranose residues with 3′, 5′ linkages, has approximately twenty-four nucleotide residues in seven turns of the helix. The helixes have the sense of a right-handed screw. The phosphate groups are closely packed about the axis of the molecule, with the pentose residues surrounding them, and the purine and pyrimidine groups projecting radially, their planes being approximately perpendicular to the molecular axis. The operation that converts one residue to the next residue in the polynucleotide chain is rotation by about 105° and translation by 3.4 Å.
This triplex model of nucleic acids, with phosphates in the center and bases on the outside, turned out to be fundamentally flawed. Yet, it played a significant role by prompting Watson and Crick in their discovery of the DNA double helix structure. While I’ve been aware of the Pauling triplex model from long ago, I had not read the original Pauling & Corey PNAS paper. Not surprisingly, I did not know what the triplex structure really looks like, other than some general ideas.
In a recent trip to Rutgers, Dr. Wilma Olson and I discussed the applications of fiber models collected in 3DNA. She drew my attention to the Pauling triplex model, and showed me Table 1 of the PNAS paper (see below), where the atomic coordinates for a nucleic acid repeating unit are listed.

The cylindrical format is the same as that for the fiber models in 3DNA. It thus seems fitting to add this historically significant triplex model to the collection. Googling revealed many interesting historical notes and comments, e.g. The Pauling-Corey Structure of DNA, and a short video Linus Pauling’s triple DNA helix model, 3D animation with basic narration. However, I failed to find a program that I can use to generate such a triplex model with generic base sequence. I decided to add the fiber --pauling option so users can easily create such a triplex model in 3D, just as they do for a classic A- and B-DNA duplex. This process has turned out to be very educational (detailed below), and the end result should be of general interest.
 in Pauling triplex")
- The left 3D image shows the nomenclature of atoms used by Pauling & Corey (see Table 1 above), which is dramatically different from current conventions. As an example, it should be the N1 atom of cytosine (a pyrimidine base), not N3, that is connected to the sugar C1′ atom in nowadays nomenclature. The corrections apply not only to base atoms, but also to the sugar and phosphate groups. The revised atom labeling (as used in the PDB) is illustrated in the 3D image on the right.
- Table 1 corresponds to the ribose sugar since it contains an O2′ atom (see also the figure above). The triplex model constructed would be RNA, but can be ‘converted’ to DNA by simply removing the O2′ atom (see below).
- Only the atomic coordinates for cytosine are listed in Table 1. The 3DNA
mutate_bases program came handy to get the corresponding atomic coordinates for A, G, T, and U. This expansion allows for the generation of Pauling’s triplex models with an arbitrary combination of the five common bases (A, C, G, T, and U).
- With the new
fiber --pauling option, now users can conveniently generate a Pauling’s triplex RNA/DNA model as shown below. Note that the one dash variant -pauling also works fine, with the additional -dna for DNA deoxyribose sugar. The PDB file (Pauling-triplex-mixed.pdb) with mixed DNA sequences can be downloaded, and the corresponding 3D image in top and side views is shown in the following figure.
fiber -pauling triplex-C10C10C10.pdb # default: 10 Cs per strand
fiber -pauling -seq=AAA triplex-A3A3A3.pdb # 3 As per strand
fiber -pauling -seq=AAAA:CCCC:GGGG Pauling-triplex-A4C4G4.pdb
fiber -pauling -seq=ACGGUU,UUGGAC,GGAACC Pauling-triplex-mixed.pdb
fiber --pauling-dna -seq=ACGGTT,TTGGAC,GGAACC Pauling-triplex-DNA.pdb

- With 3DNA’s
find_pair/analyze pair of programs, one can get the structural parameters corresponding to the Pauling triplex model. Not surprising, the repeating dinucleotide along each strand has a twist of 105°, and a rise of 3.4 Å. Notably, the sugar has a C2′-endo conformation.
3DNA contains 55 fiber models compiled from literature, plus a derived RNA model (as of v2.1). To the best of my knowledge, this is the most comprehensive collection of regular DNA/RNA models. Please see Table 4 of the 2003 3DNA NAR paper for detailed structural features of these models and references.
The 55 models are based on the following works:
- Chandrasekaran & Arnott (from #1 to #43) — the most well-known set of fiber models
- Alexeev et al. (#44-#45)
- van Dam & Levitt (#46-#47)
- Premilat & Albiser (#48-#55)
The utility program fiber makes the generation of all these fiber models in a simple, consistent interface, and produces coordinate files in either PDB or PDBML format. Of those models, some can be built with an arbitrary sequence of A, C, G and T (e.g., A-/B-/C-DNA from calf thymus), while others are of fixed sequences (e.g., Z-DNA with GC repeats). The sequence can be specified either from command-line or a plain text file, in either lower, UPPER, or MixED cases.
Once 3DNA in properly installed, the command-line interface is the most versatile and convenient way to generate, e.g., a regular double-stranded DNA (mostly, B-DNA) of arbitrary sequence. The command-help message (generated with fiber -h) is as below:
NAME
fiber - generate 55 fiber models based on Arnott and other's work
SYNOPSIS
fiber [OPTION] PDBFILE
DESCRIPTION
generate 55 fiber models based on the repeating unit from Arnott's
work, including the canonical A-, B-, C- and Z-DNA, triplex, etc
-xml output structure coordinates in PDBML format
-num a structure identification number in the range (1-55)
-m, -l brief description of the 55 fiber structures
-a, -1 A-DNA model (calf thymus)
-b, -4 B-DNA (calf thymus, default)
-c, -47 C-DNA (BII-type nucleotides)
-d, -48 D(A)-DNA ploy d(AT) : ploy d(AT) (right-handed)
-z, -15 Z-DNA poly d(GC) : poly d(GC)
-rna for RNA with arbitrary base sequence
-seq=string specifying an arbitrary base sequence
-single output a single-stranded structure
-h this help message (any non-recognized options will do)
INPUT
An structural identification number (symbol)
EXAMPLES
fiber fiber-BDNA.pdb
# fiber -4 fiber-BDNA.pdb
# fiber -b fiber-BDNA.pdb
fiber -a fiber-ADNA.pdb
fiber -seq=AAAGGUUU -rna fiber-RNA.pdb
fiber -seq=AAAGGUUU -rna -single fiber-ssRNA.pdb
OUTPUT
PDB file
SEE ALSO
analyze, anyhelix, find_pair
AUTHOR
3DNA v2.3-2016sept06, created and maintained by Xiang-Jun Lu (PhD)
Please post questions/comments on the 3DNA Forum: http://forum.x3dna.org/
Moreover, the w3DNA, 3D-DART web-interfaces, and the PyMOL wrapper make it easy to generate a regular DNA (or RNA) model, especially for occasional users or for educational purposes.
In principle, nothing is worth showing off with regard to 3DNA’s fiber model generation functionality. Nevertheless, this handy tool serves as a clear example of the differences between a “proof of concept” and a pragmatic software application. I initially decided to work on this tool simply for my own convenience. At that time, I had access to A-DNA and B-DNA fiber model generators, each as a separate program. Moreover, the constructed models did not comply to the PDB format in atom naming, among other subtitles.
I started with the Chandrasekaran & Arnott fiber models which I had a copy of data files. However, there were many details to work out, typos to correct, etc. to put them in a consistent framework. For other models, I had to read each original publication, and to type raw atomic cylindrical coordinates into computer. Again, quite a few inconsistencies popped up between the different publications with a time span over decades.
Overall, it was a quite tedious undertaking, requiring great attention to details. I am glad that I did that: I learned so much from the process, and more importantly, others can benefit from my effort. As I put in the 3DNA Nature Protocol paper (BOX 6 | FIBER-DIFFRACTION MODELS),
In preparing this set of fiber models, we have taken great care to ensure the accuracy and consistency of the models. For completeness and user verification, 3DNA includes, in addition to 3DNA-processed files, the original coordinates collected from the literature.
For those who want to understand what’s going on under the hood, there is no better way than to try to reproduce the process using, e.g., fiber B-DNA as an example.
From the very beginning, I had expected the 3DNA fiber functionality to serve as a handy tool for building a regular DNA duplex of chosen sequence. Over the years, the fiber program has gradually attracted attention from the community. The recent PyMOL wrapper by Thomas Holder is a clear sign of its increased popularity, and has prompted me to write this post, adapted largely from the one titled Fiber models in 3DNA make it easy to build regular DNA helices (dated Friday, October 9, 2009).
See also PyMOL wrapper to 3DNA fiber models

Given below is the content of the README file for fiber models in 3DNA:
1. The repeating units of each fiber structure are mostly based on the
work of Chandrasekaran & Arnott (from #1 to #43). More recent fiber
models are based on Alexeev et al. (#44-#45), van Dam & Levitt (#46
-#47) and Premilat & Albiser (#48-#55).
2. Clean up of each residue
a. currently ignore hydrogen atoms [can be easily added]
b. change ME/C7 group of thymine to C5M
c. re-assign O3' atom to be attached with C3'
d. change distance unit from nm to A [most of the entries]
e. re-ordering atoms according to the NDB convention
3. Fix up of problem structures.
a. str#8 has no N9 atom for guanine
b. str#10 is not available from the disk, manually input
c. str#14 C5M atom was named C5 for Thymine, resulting two C5 atoms
d. str#17 has wrong assignment of O3' atom on Guanine
e. str#33 has wrong C6 position in U3
f. str#37 to #str41 were typed in manually following Arnott's
new list as given in "Oxford Handbook of Nucleic Acid Structure"
edited by S. Neidle (Oxford Press, 1999)
g. str#38 coordinates for N6(A) and N3(T) are WRONG as given in the
original literature
h. str#39 and #40 have the same O3' coordinates for the 2nd strand
4. str#44 & 45 have fixed strand II residues (T)
5. str#46 & 47 have +z-axis upwards (based on BI.pdb & BII.pdb)
6. str#48 to 55 have +z-axis upwards
List of 55 fiber structures
id# Twist Rise Structure description
(dgrees) (A)
-------------------------------------------------------------------------------
1 32.7 2.548 A-DNA (calf thymus; generic sequence: A, C, G and T)
2 65.5 5.095 A-DNA poly d(ABr5U) : poly d(ABr5U)
3 0.0 28.030 A-DNA (calf thymus) poly d(A1T2C3G4G5A6A7T8G9G10T11) :
poly d(A1C2C3A4T5T6C7C8G9A10T11)
4 36.0 3.375 B-DNA (calf thymus; generic sequence: A, C, G and T)
5 72.0 6.720 B-DNA poly d(CG) : poly d(CG)
6 180.0 16.864 B-DNA (calf thymus) poly d(C1C2C3C4C5) : poly d(G6G7G8G9G10)
7 38.6 3.310 C-DNA (calf thymus; generic sequence: A, C, G and T)
8 40.0 3.312 C-DNA poly d(GGT) : poly d(ACC)
9 120.0 9.937 C-DNA poly d(G1G2T3) : poly d(A4C5C6)
10 80.0 6.467 C-DNA poly d(AG) : poly d(CT)
11 80.0 6.467 C-DNA poly d(A1G2) : poly d(C3T4)
12 45.0 3.013 D-DNA poly d(AAT) : poly d(ATT)
13 90.0 6.125 D-DNA poly d(CI) : poly d(CI)
14 -90.0 18.500 D-DNA poly d(A1T2A3T4A5T6) : poly d(A1T2A3T4A5T6)
15 -60.0 7.250 Z-DNA poly d(GC) : poly d(GC)
16 -51.4 7.571 Z-DNA poly d(As4T) : poly d(As4T)
17 0.0 10.200 L-DNA (calf thymus) poly d(GC) : poly d(GC)
18 36.0 3.230 B'-DNA alpha poly d(A) : poly d(T) (H-DNA)
19 36.0 3.233 B'-DNA beta2 poly d(A) : poly d(T) (H-DNA beta)
20 32.7 2.812 A-RNA poly (A) : poly (U)
21 30.0 3.000 A'-RNA poly (I) : poly (C)
22 32.7 2.560 Hybrid poly (A) : poly d(T)
23 32.0 2.780 Hybrid poly d(G) : poly (C)
24 36.0 3.130 Hybrid poly d(I) : poly (C)
25 32.7 3.060 Hybrid poly d(A) : poly (U)
26 36.0 3.010 10-fold poly (X) : poly (X)
27 32.7 2.518 11-fold poly (X) : poly (X)
28 32.7 2.596 Poly (s2U) : poly (s2U) (symmetric base-pair)
29 32.7 2.596 Poly (s2U) : poly (s2U) (asymmetric base-pair)
30 32.7 3.160 Poly d(C) : poly d(I) : poly d(C)
31 30.0 3.260 Poly d(T) : poly d(A) : poly d(T)
32 32.7 3.040 Poly (U) : poly (A) : poly(U) (11-fold)
33 30.0 3.040 Poly (U) : poly (A) : poly(U) (12-fold)
34 30.0 3.290 Poly (I) : poly (A) : poly(I)
35 31.3 3.410 Poly (I) : poly (I) : poly(I) : poly(I)
36 60.0 3.155 Poly (C) or poly (mC) or poly (eC)
37 36.0 3.200 B'-DNA beta2 Poly d(A) : poly d(U)
38 36.0 3.240 B'-DNA beta1 Poly d(A) : poly d(T)
39 72.0 6.480 B'-DNA beta2 Poly d(AI) : poly d(CT)
40 72.0 6.460 B'-DNA beta1 Poly d(AI) : poly d(CT)
41 144.0 13.540 B'-DNA Poly d(AATT) : poly d(AATT)
42 32.7 3.040 Poly(U) : poly d(A) : poly(U) [cf. #32]
43 36.0 3.200 Beta Poly d(A) : Poly d(U) [cf. #37]
44 36.0 3.233 Poly d(A) : poly d(T) (Ca salt)
45 36.0 3.233 Poly d(A) : poly d(T) (Na salt)
46 36.0 3.38 B-DNA (BI-type nucleotides; generic sequence: A, C, G and T)
47 40.0 3.32 C-DNA (BII-type nucleotides; generic sequence: A, C, G and T)
48 87.8 6.02 D(A)-DNA ploy d(AT) : ploy d(AT) (right-handed)
49 60.0 7.20 S-DNA ploy d(CG) : poly d(CG) (C_BG_A, right-handed)
50 60.0 7.20 S-DNA ploy d(GC) : poly d(GC) (C_AG_B, right-handed)
51 31.6 3.22 B*-DNA poly d(A) : poly d(T)
52 90.0 6.06 D(B)-DNA poly d(AT) : poly d(AT) [cf. #48]
53 -38.7 3.29 C-DNA (generic sequence: A, C, G and T) (depreciated)
54 32.73 2.56 A-DNA (generic sequence: A, C, G and T) [cf. #1]
55 36.0 3.39 B-DNA (generic sequence: A, C, G and T) [cf. #4]
-------------------------------------------------------------------------------
List 1-41 based on Struther Arnott: ``Polynucleotide secondary structures:
an historical perspective'', pp. 1-38 in ``Oxford Handbook of Nucleic
Acid Structure'' edited by Stephen Neidle (Oxford Press, 1999).
#42 and #43 are from Chandrasekaran & Arnott: "The Structures of DNA
and RNA Helices in Oriented Fibers", pp 31-170 in "Landolt-Bornstein
Numerical Data and Functional Relationships in Science and Technology"
edited by W. Saenger (Springer-Verlag, 1990).
#44-#45 based on Alexeev et al., ``The structure of poly(dA) . poly(dT)
as revealed by an X-ray fiber diffraction''. J. Biomol. Str. Dyn, 4,
pp. 989-1011, 1987.
#46-#47 based on van Dam & Levitt, ``BII nucleotides in the B and C forms
of natural-sequence polymeric DNA: a new model for the C form of DNA''.
J. Mol. Biol., 304, pp. 541-561, 2000.
#48-#55 based on Premilat & Albiser, ``A new D-DNA form of poly(dA-dT) .
poly(dA-dT): an A-DNA type structure with reversed Hoogsteen Pairing''.
Eur. Biophys. J., 30, pp. 404-410, 2001 (and several other publications).
{kind=link}