[Job] Staff Associate II (Computational Structural Biology) at Columbia University
The X3DNA-DSSR resource is at the forefront of structural bioinformatics, developing advanced tools for analyzing and modeling nucleic acid structures. We are seeking a highly motivated Staff Associate II to join our team and contribute to our next-generation analysis and visualization engine.
To see our resource in action, please visit wDSSR, our new web interface for dissecting and modeling 3D nucleic acid structures: https://web.x3dna-dssr.org/.
We are looking for a candidate with a strong scientific background in structural biology or bioinformatics and a desire to contribute to peer-reviewed publications through community-driven data analysis. We value individuals who are eager to learn, adapt to new technical challenges, and support the global research community.
For the full job description and to submit your application, please visit the official Columbia University posting:
https://apply.interfolio.com/183705
Announcing wDSSR: The Next-Generation Web Interface to X3DNA-DSSR
Dear 3DNA/DSSR Community,
We are thrilled to announce the official launch of wDSSR (https://web.x3dna-dssr.org/), the powerful new web interface to the X3DNA-DSSR analytical engine.
Developed by Drs. Shuxiang Li and Xiang-Jun Lu and supported by NIH grant R24GM153869, wDSSR represents a major leap forward from our highly popular 2019 Web 3DNA 2.0 framework. While Web 3DNA 2.0 has faithfully served the community for the analysis, visualization, and modeling of 3D nucleic acid structures, wDSSR was built from the ground up to take full advantage of modern web technologies and the latest DSSR backend capabilities.
A Modern, Streamlined Scientific Workflow
We have completely overhauled the user interface to provide a clean, intuitive, and task-driven experience. The core modeling and analysis tools are now seamlessly organized into a logical, single-word scientific workflow: Analyze, Rebuild, Model, Circularize, Mutate, Assemble, and Visualize.
Spotlight Feature: The "Assemble" Module
One of the most exciting upgrades is the newly renamed Assemble tab (formerly "Composite"). This advanced composite model builder allows you to effortlessly construct complex, higher-order models by linking any combination of nucleic acid duplexes or protein-DNA/RNA complexes. You can quickly connect up to six distinct target structures, ranging from simple linked A-DNA and B-DNA duplexes to large, protein-decorated structural assemblies.
Immediate Global Adoption
Although wDSSR has just launched, we are incredibly humbled to share that it is already seeing rapid worldwide adoption! According to recent network infrastructure data, the new interface is actively being used by researchers across North America, South America, Europe, and Asia. Within just a few days, we have recorded active sessions from prestigious institutions around the globe, including:
- The Weizmann Institute of Science in Israel
- Katholieke Universiteit Leuven in Belgium
- Queen's University in Canada
- Universidad Nacional Autonoma de Mexico (UNAM) in Mexico
- Emory University and the Wadsworth Centers Laboratories and Research in the United States
- Jawaharlal Nehru University and the China Education and Research Network in Asia
How to Cite
While a dedicated paper for wDSSR is currently in preparation, researchers should cite the server using its URL (https://web.x3dna-dssr.org/) alongside the 2019 Web 3DNA 2.0 paper and the foundational 2015 DSSR paper. Full details and funding acknowledgements can be found on our newly consolidated About page.
We invite you all to try out the new wDSSR platform! As always, your feedback is invaluable to us, and we encourage you to share your thoughts, questions, and structural models via the newly updated Questions & Feedback link in the wDSSR footer.
Happy modeling!
Over the years, the fiber utility program has become a handy way to generate standard B-DNA and A-DNA structures, as evident from citations to 3DNA. Nevertheless, the currently collected 55 experimental fiber models, comprehensive as they are, do not include one for canonical double-stranded (ds) RNA or single-stranded (ss) RNA structures of generic A/C/G/U sequence.
This situation is best illustrated by a recent article by Charles Brooks and Hashim Al-Hashimi and their co-workers, titled Unraveling the structural complexity in a single-stranded RNA tail: implications for efficient ligand binding in the prequeuosine riboswitch [Nucleic Acids Research, 40(3) 1345–1355 (2012)] , where they wrote:
Idealized A-form structures were constructed using Insight II (Molecular Simulations, Inc.) correcting the propeller twist angles from +15° to –15° using an in-house program, as previously described (47). The complementary strand was removed and the resulting ssRNA used in NMR data analysis. B-form helices were constructed using W3DNA (48).
As of 3DNA v2.1, however, that’s no longer the case: now the fiber utility provides direct support for generating idealized dsRNA or ssRNA structures of arbitrary A/C/G/U sequence. As always, the new functionality can be best illustrated with examples. Let’s build ssRNAs of the wild-type (5’-AUAAAAAACUAA-3’) and A29C mutated form (5’-AUAACAAACUAA-3’) used in the work cited above:
fiber -r -s -seq=AUAAAAAACUAA wt-12nt.pdb
fiber -r -s -seq=AUAACAAACUAA mt-12nt.pdb
Here the -r option is for RNA, -s for a ss structure, and -seq for the specific base sequence. The generated ssRNA structure for the wild-type sequence is named wt-12nt.pdb, and that for the mutated sequence named mt-12nt.pdb.
Note that the new RNA model is based on Struther Arnott’s work of fiber A-DNA from calf thymus (#1 in the list). The dsRNA, as its dsDNA counterpart, has a helical twist of 32.7° and a helical rise of 2.548 Å. Relevant to the above citation, here the propeller twist angle of each base pair is –10.5°, a negative value similar to that observed in high-resolution x-ray crystal structures. Furthermore, you can easily verify the three numbers with the following commands:
fiber -r -seq=AUAAAAAACUAA wt-12nt.pdb
find_pair wt-12nt.pdb stdout | analyze stdin
In summary, it is very easy to generate canonical RNA structures with the revised fiber command. Through its integrated analysis routine, 3DNA can also be used to check structural features of the resultant RNA models. Moreover, as mentioned in the opening post What can 3DNA do for RNA structures? on the forum, 3DNA has much to offer in the filed of RNA structural bioinformatics.

At the C2B2 party this afternoon, I was asked the question: “Does 3DNA work for RNA?” Well, a good question, indeed. The short answer is definitely, YES. However, a detailed explanation is needed to address the underlying intuitive assumption: 3DNA is only for DNA.
- The name 3DNA was due to Dr. Olson, after we struggled quite a while. Initially, we played with NuStar (which was actually cited once by Richard Dickerson et al.), and Carnival etc. I still remember the day when Dr. Olson asked me “How about 3DNA?” We immediately reached an agreement: that’s it — what a cute name! Another advantage (as it becomes clear later): since 3DNA starts with ‘3’, it (mostly) shows up right at the top of many on-line lists of bioinformatics tools.
- Interpreted literally, 3DNA could mean 3-DNA, i.e., the three most common types of DNA: A-, B- and Z-form. That may be one of the reasons where the misconception that 3DNA is only for 3DNA comes from. Another reason could be that structural work on DNA is what the Olson lab best known for.
- The number ‘3’ in 3DNA should also be associated with its three key components: analysis, rebuilding and visualization. In a sense, this is my favorite.
- Of course, 3DNA stands for 3D-NA, 3-Dimensional Nucleic Acids, as expressed explicitly in the titles of our two 3DNA papers (2003 NAR and 2008 NP).
The applications of 3DNA to RNA structures can be broadly categorized as follows:
- Automatically detect all existing base-pairs, Watson-Crick (A-U, G-C, wobble G-U) or non-canonical, using a set of simple geometric criteria. Furthermore, it has a unique base-pair classification system based on the six numerical structural parameters, suitable for database storage and search.
- Automatically detect all triplets or higher-order base-associations.
- Automatically detect double helical regions, regardless of backbone connection, thus ideal for finding pseudo-continuous coaxial stacking.
- The above three features are seamlessly integrated with the visualization component to allow for easy generation of publication quality images. See the 3DNA 2008 NP paper for detailed examples.
As further examples, the following two RNA publications take advantage of find_pair from 3DNA:
It is well worth noting that the base-pair detecting algorithm in RNAView is based on an earlier version of find_pair, a basic fact ignored in the RNAView publication.
In summary, 3DNA works for RNA as well as for DNA, and more.
 A video overview of DSSR
A video overview of DSSR
DSSR (Dissecting the Spatial Structure of RNA) is an integrated software tool for the analysis/annotation, model building, and schematic visualization of 3D nucleic acid structures (see the figures below and the video overview). It is built upon the well-known, tested, and trusted 3DNA suite of programs. DSSR has been made possible by the developer’s extensive user-support experience, detail-oriented software engineering skills, and expert domain knowledge accumulated over two decades. It streamlines tasks in RNA/DNA structural bioinformatics, and outperforms its ‘competitors’ by far in terms of functionality, usability, and support.
Wide citations. DSSR has been widely cited in scientific literature, including: (i) “Selective small-molecule inhibition of an RNA structural element” (Nature, 2015; Merck Research Laboratories), (ii) “The structure of the yeast mitochondrial ribosome” (Science, 2017), (iii) “RNA force field with accuracy comparable to state-of-the-art protein force fields” (PNAS, 2018; D. E. Shaw Research), (iv) “Predicting site-binding modes of ions and water to nucleic acids using molecular solvation theory” (JACS, 2019), (v) “RIC-seq for global in situ profiling of RNA-RNA spatial interactions” (Nature, 2020), and (vi) “DNA mismatches reveal conformational penalties in protein-DNA recognition” (Nature, 2020).
Broad integrations. To make DSSR as widely accessible as possible, I have initiated collaborations with the principal developers of Jmol and PyMOL. The DSSR-Jmol and DSSR-PyMOL integrations bring unparalleled search capabilities (e.g., ‘select junctions’ for all multi-branch loops) and innovative visualization styles into 3D nucleic acid structures. DSSR has also been adopted into numerous other structural bioinformatics resources, including: (i) URS, (ii) RiboSketch, (iii) RNApdbee, (iv) forgi, (v) RNAvista, (vi) VeriNA3d, (vii) RNAMake, (viii) ElTetrado, (ix) DNAproDB, (x) LocalSTAR3D, (xi) IPANEMAP, and (xii) RNANet.
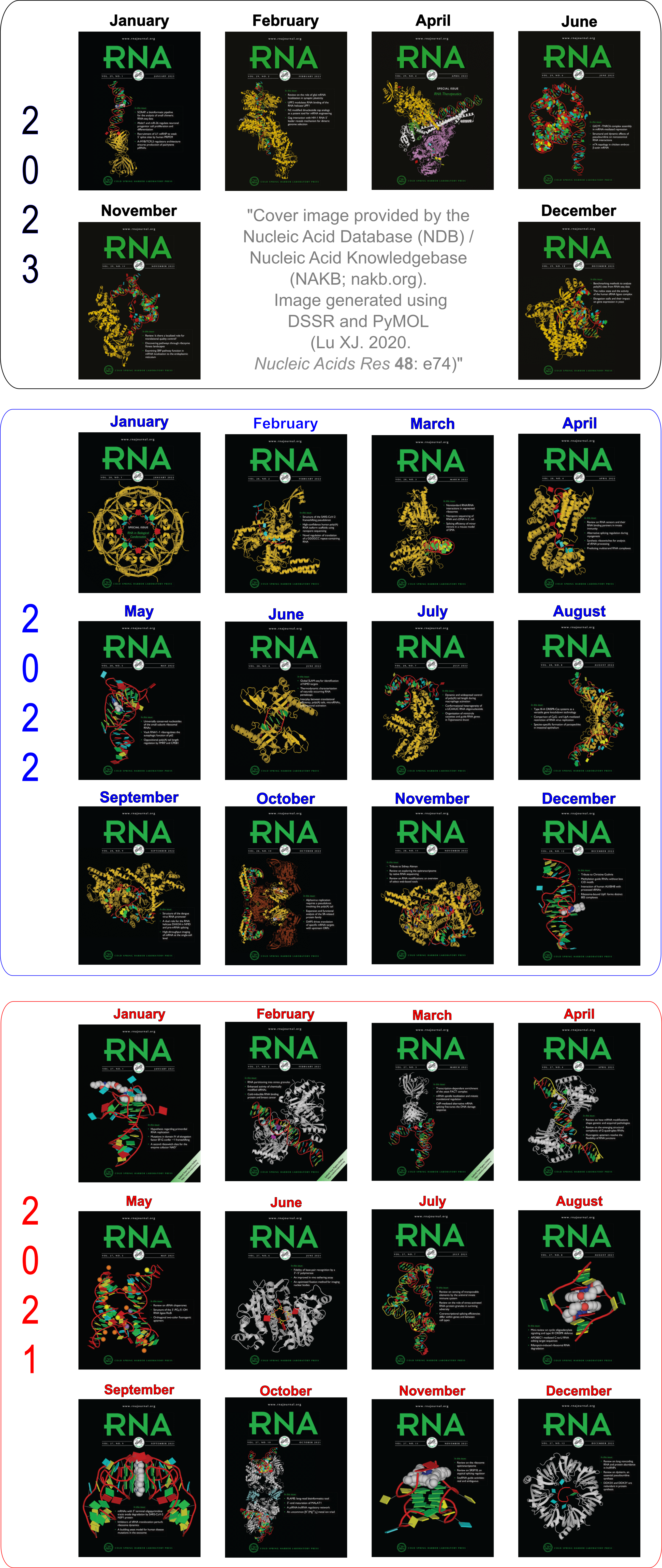
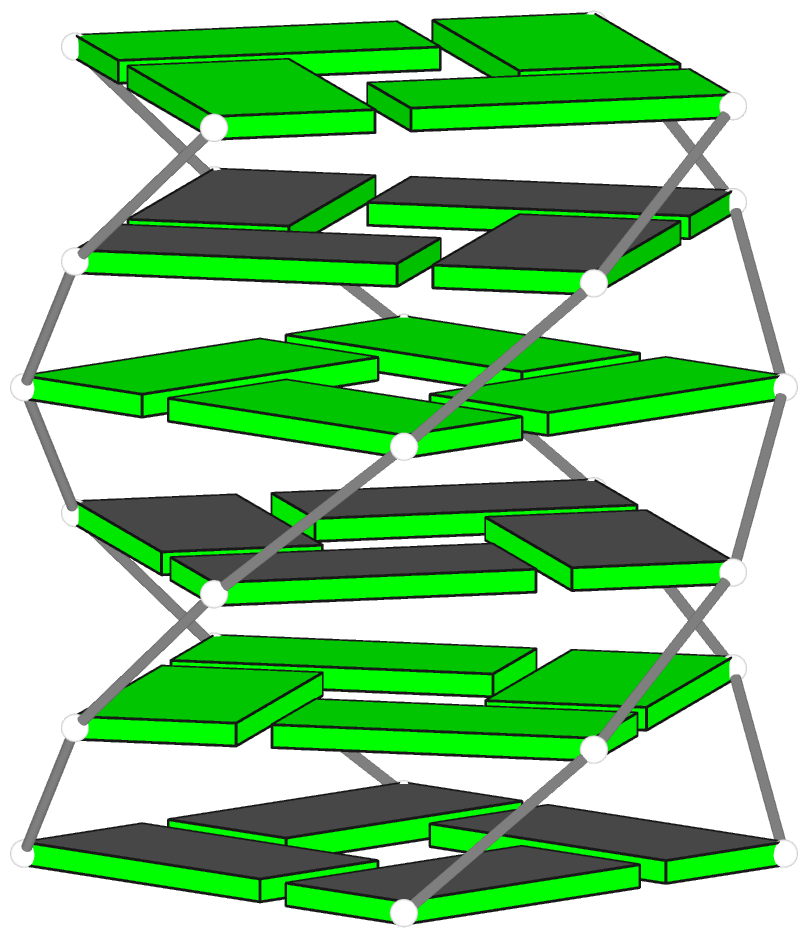
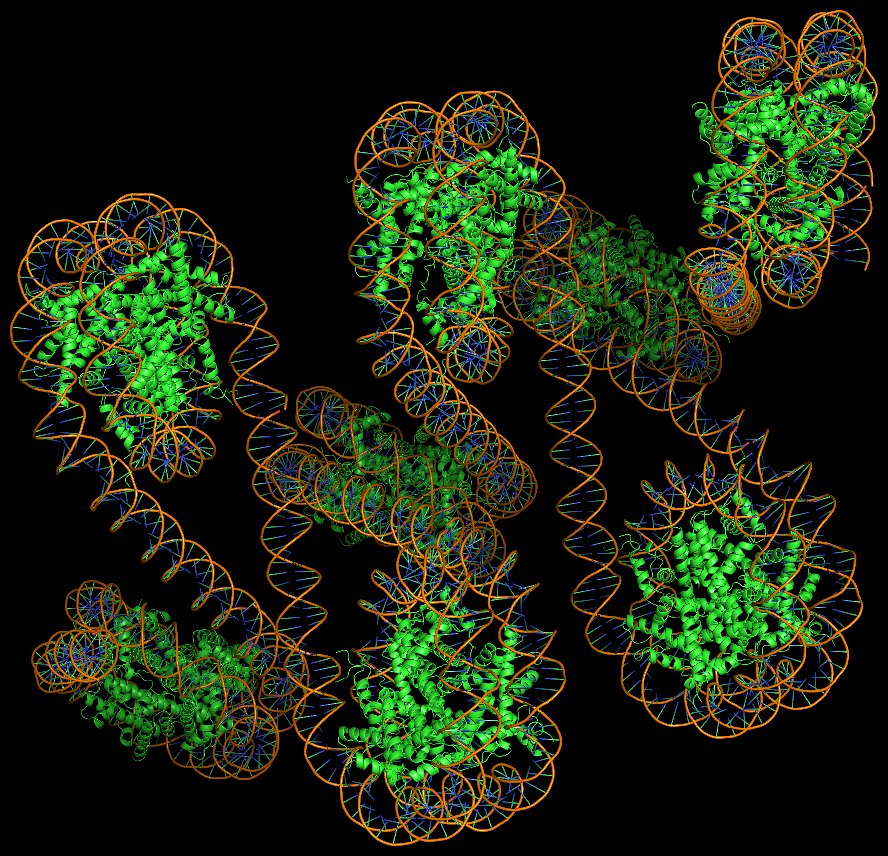
Advanced features. DSSR may be licensed from Columbia University. DSSR Pro is the commercial version. It has more functionalities than DSSR basic (the free academic version), including: (i) homology modeling via in silico base mutations, a feature employed by Merck scientists, (ii) easy generation of regular helical models, including circular or super-helical DNA (see figures below), (iii) creation of customized structures with user-specified base sequences and rigid-body parameters, (iv) efficient processing of molecular dynamics (MD) trajectories, (v) detailed characterization of DNA-protein or RNA-protein spatial interactions, and (vi) template-based modeling of DNA-protein complexes (see figures below). DSSR Pro supersedes 3DNA. It integrates the disparate analysis and modeling programs of 3DNA under one umbrella, and offers new advanced features, through a convenient interface. For example, with the mutate module of DSSR Pro, one can automatically perform the following tasks: (i) mutate all bases to Us, (ii) mutate bases in hairpin loops to Gs, and (iii) mutate G–C Watson-Crick pairs to C–G, and A–U to U–A. Moreover, DSSR Pro includes an in-depth user manual and one-year technical support from the developer.
Quality control. DSSR is a solid software product that excels in RNA structural bioinformatics. It is written in strict ANSI C, as a single command-line program. It is self-contained, with zero runtime dependencies on third-party libraries. The binary executables for macOS, Linux, and Windows are just ~2MB. DSSR has been extensively tested using all nucleic-acid-containing structures in the PDB. It is also routinely checked with Valgrind to avoid memory leaks. DSSR requires no set up or configuration: it simply works.

Theoretical models of G-quadruplexes, created using DSSR Pro.

Template-based modeling of DNA-protein complexes using DSSR Pro.
Here are two chromatin-like models using PDB entry 4xzq as the template.


Circular DNA duplexes modeled using DSSR Pro.
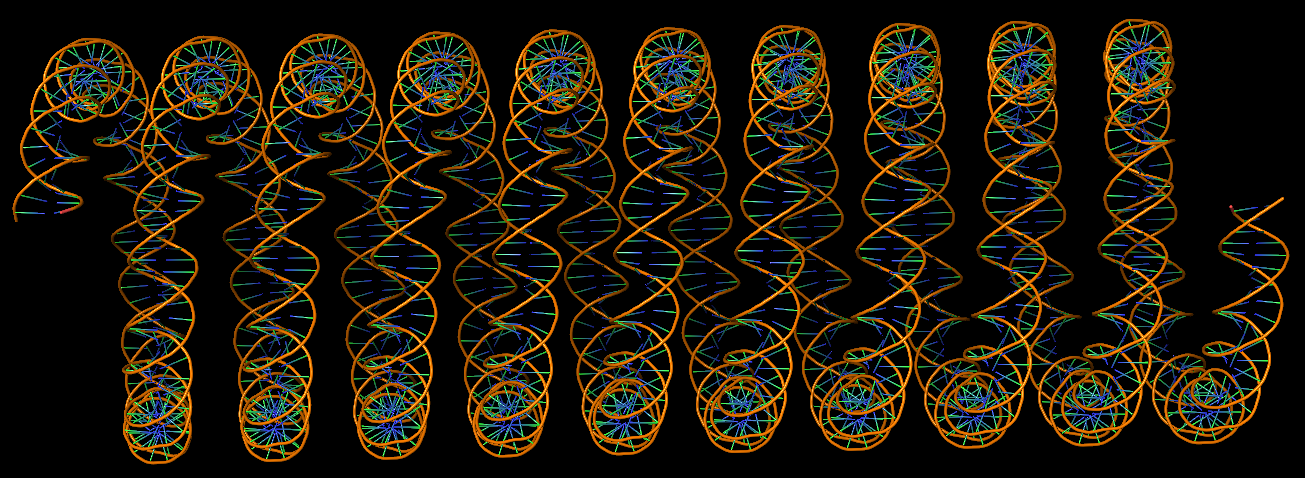
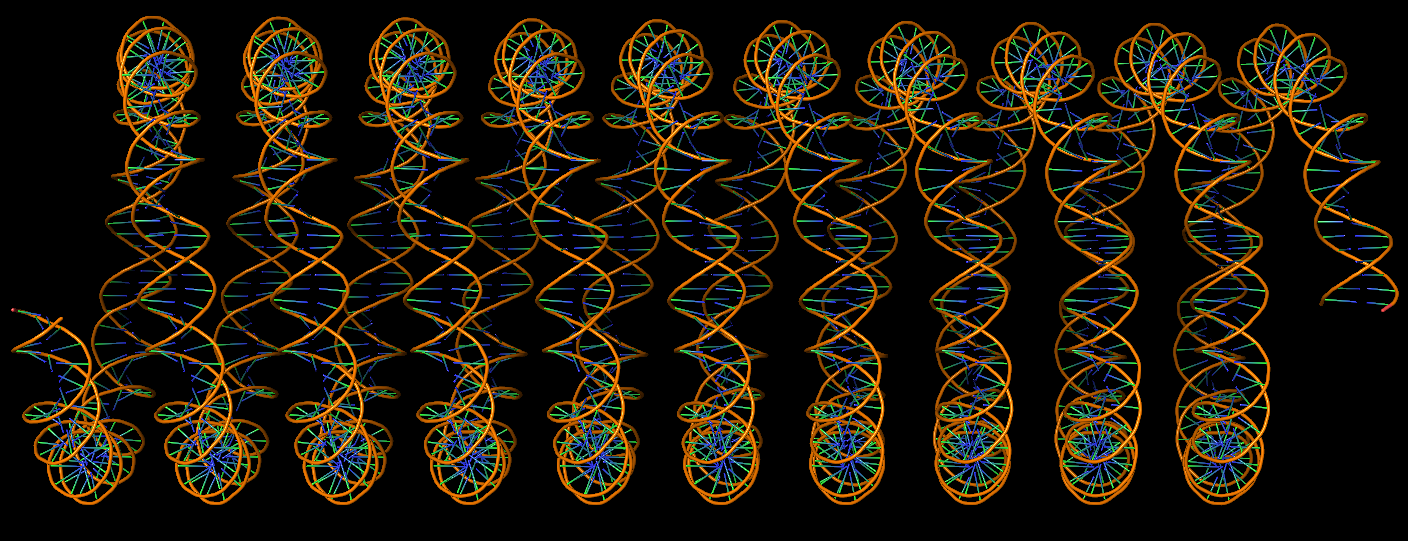
DNA super helices modeled using DSSR Pro.

Innovative cartoon-block schematics enabled by the DSSR-PyMOL integration for six representative PDB entries. Watson-Crick pairs are shown as long blocks with minor-groove edges in black (A, B), G-tetrads represented as square blocks and the metal ion as sphere ©, the ligand rendered as balls-and-sticks (D), and proteins depicted as purple cartoons (E, F). Color code for base blocks: A, red; C, yellow; G, green; T, blue; U, cyan; G-tetrad, green; WC-pairs, per base in the leading strand. Visit http://skmatic.x3dna.org.
Recommended in Faculty Opinions: “simple and effective”, “Good for Teaching”.
Employed by the NDB to create cover images of the RNA Journal.
The following links point to tools that are relevant to 3DNA.
- Curves+ — an updated version of the well-known Curves program, and it conforms to the standard base reference frame.
- 3D-DART — 3DNA-Driven DNA Analysis and Rebuilding Tool. Another web-interface to commonly used 3DNA functionality.
- do_x3dna — “do_x3dna has been developed for analysis of the DNA/RNA dynamics during the molecular dynamics simulations. It uses the 3DNA package to calculate several structural descriptors of DNA/RNA from the GROMACS MD trajectory. It executes 3DNA tools to calculate these descriptors and subsequently, extracts these output and saves into external output files as a function of time.”
- SwS — a Solvation web Service for Nucleic Acids where 3DNA plays a role.
- Raster3D — a set of tools for generating high-quality raster images of proteins or other molecules.
- MolScript — a program for displaying molecular 3D structures, such as proteins, in both schematic and detailed representations.
- Jmol — an open-source Java viewer for chemical structures in 3D with features for chemicals, crystals, materials, and biomolecules.
- PyMOL — a user-sponsored molecular visualization system on an open-source foundation.
- ImageMagick — a software suite to create, edit, compose, or convert bitmap images.
- NDB — Nucleic acids database.
- SBGrid — Excellent services for structural biology laboratories as well software developers.
The v2.1 release of 3DNA, currently in beta, contains many refinements of existing C programs, a complete migration from Perl scripts to Ruby, and additions of several significant new programs. All know bugs in v2.0 have been fixed. Highlights include:
- Added mutate_bases to perform in silico base mutations in nucleic-acid-containing structures (DNA, RNA, and their complexes with ligands and proteins). The program has two key and unique features: (1) the sugar-phosphate backbone conformation is untouched; (2) the base reference frame (position and orientation) is reserved, i.e., the mutated structure shares the same base-pair/step parameters as those of the native structure.
- Added
x3dna_ensemble, a Ruby script to automate the processing of an NMR structure ensemble or MD trajectories in MODEL/ENDMDL delineated PDB format. It has sub-commands analyze, extract, reorient, and block_iamge. To add: convert to transform Amber, Gromacs or CHARMM trajectories.
- Enhanced
find_pair with -c+ option for generating input to Curves+.
- Expanded
fiber with the -s option for generating single stranded structures; the -seq option for specifying base sequence directly on the command line; and the -r option for generating RNA structures (single or double stranded) of arbitrary ACGU sequences.
- Updated the ‘baselist.dat’ file to incorporate all types of NDB/PDB nucleotides as of February 15, 2015; refined
find_pair/analyze/mutate_bases etc to automatically detect and assign of modified bases.
- Renamed Atomic_a.pdb and Atomic.a.pdb etc for modified bases to account for Mac OS X filesystem case sensitivity issue; Copied all Perl scripts to a new directory
perl_scripts/.
- 3DNA now generates PDB files that are compliant with PDB format v3.x, and also has option to allow for three-letter nucleotide names, thus directly compatible with PdbViewer and HADDock. An option is provided to convert 3DNA-generated base rectangular blocks in Alchemy to the more widely accepted MDL molfile format (e.g. by PyMOL).
Recently, I (together with Drs. Wilma Olson and Harmen Bussemaker – a team with a unique combination of complementary expertise) published a new article in Nucleic Acids Research (NAR): The RNA backbone plays a crucial role in mediating the intrinsic stability of the GpU dinucleotide platform and the GpUpA/GpA mini duplex. The key findings of this work are summarized in the abstract:
The side-by-side interactions of nucleobases contribute to the organization of RNA, forming the planar building blocks of helices and mediating chain folding. Dinucleotide platforms, formed by side-by-side pairing of adjacent bases, frequently anchor helices against loops. Surprisingly, GpU steps account for over half of the dinucleotide platforms observed in RNA-containing structures. Why GpU should stand out from other dinucleotides in this respect is not clear from the single well-characterized H-bond found between the guanine N2 and the uracil O4 groups. Here, we describe how an RNA-specific H-bond between O2’(G) and O2P(U) adds to the stability of the GpU platform. Moreover, we show how this pair of oxygen atoms forms an out-of-plane backbone ‘edge’ that is specifically recognized by a non-adjacent guanine in over 90% of the cases, leading to the formation of an asymmetric miniduplex consisting of ‘complementary’ GpUpA and GpA subunits. Together, these five nucleotides constitute the conserved core of the well-known loop-E motif. The backbone-mediated intrinsic stabilities of the GpU dinucleotide platform and the GpUpA/GpA miniduplex plausibly underlie observed evolutionary constraints on base identity. We propose that they may also provide a reason for the extreme conservation of GpU observed at most 5’-splice sites.
As a nice surprise, this publication was selected by NAR as a featured article! According to the NAR website:
Featured Articles highlight the best papers published in NAR. These articles are chosen by the Executive Editors on the recommendation of Editorial Board Members and Referees. They represent the top 5% of papers in terms of originality, significance and scientific excellence.
I feel very gratified with the “extra” recognition. From my own perspective, I can easily rank this paper as the top one in my publication list: from the very beginning, I has been struck by the simplicity and elegance of the GpU story. Hopefully, time will verify the validity of this scientific contribution.
Behind the hood, though, there is a long, complex (sometimes perplexing), yet interesting story associated with this work. Here is how it got started. While writing the 3DNA 2008 Nature Protocols (NP) paper, I selected the (previously undocumented) ‘-p’ option of find_pair to showcase its capability to identify higher-order base associations, using the large ribosomal subunit (1jj2) as an example. I noticed the unexpected O2’(G)⋅⋅⋅O2P H-bond within the GpU dinucleotide platform in a pentaplet (Figure A below). I was/am well aware of Leontis-Westholf’s pioneering work on Geometric nomenclature and classification of RNA base pairs which involves three distinct edges – the Watson-Crick edge, the Hoogsteen edge, and the Sugar edge, yet without taking into consideration of possible sugar-phosphate backbone interactions (Figure B below). So I decided to double-check, just to be sure that the H-bond was not spurious due to defects in the H-bond detecting scheme of find_pair, and the finding was very surprising.

The following section was re-added into the 3DNA NP paper in the very last revision:
It is also worth noting that the G1971–U1972 platform is stabilized not only by the well-characterized G(N2)⋅⋅⋅U(O4) H-bond interaction, but also by a little-noticed G(O2’)⋅⋅⋅U(O2P) sugar-phosphate backbone interaction (Fig. 6a). Examination of the 50S large ribosomal unit (1JJ2) alone reveals ten such double H-bonded G–U platforms, far more occurrences than those registered by any other dinucleotide platform (including A–A) in this structure. Apparently, the G–U platform is more stable than other platforms with only a single base–base H-bond interaction. We are currently investigating this overrepresented G–U dinucleotide platform in other RNA structures. (p.1226)
See also Is the O2’(G)…O2P H-bond in GpU platforms real?
Structural analysis of nucleic acids used to be a rather tedious process, especially for irregular, complicated RNA structures and nucleic-acid/protein complexes [e.g., the large ribosomal subunit of H. marismortui (1jj2)]. Without valid base-pairing information arranged properly in a duplex fragment as input, analysis programs such as Curves+ and analyze/cehs in 3DNA would produce meaningless results. The program find_pair in 3DNA was originally created to solve this specific problem, i.e., to generate an input file to 3DNA analysis routines directly from a nucleic-acid containing structure in PDB format. It is what makes nucleic acids structural analysis a routine process — running through thousands of structures from NDB/PDB can be fully automated.
Overall, find_pair has more than fulfilled the goal of its initial design (as stated above). Over the past few years, its functionality has been expanded and continuously refined (kaizen 改善), making find_pair itself a full-featured application. Now, it is efficient, robust, and its simple command line interface allows for easy integration with other bioinformatics tools. Properly acknowledged or otherwise, find_pair has served (at least) as one of the key components in many other applications (RNAView, BPS, SwS, ARTS, to name just a few). Indeed, find_pair is by far the single program in 3DNA that has received the most questions (as evident from the 3DNA forum).
While I still have to write a method paper to describe the underlying algorithms of find_pair in detail — i.e., for identifying nucleotides, H-bonds, base pairs, high-order base associations, and double helical regions — the basic idea is intuitive and very easy to understand: as summarized in our recent GpU paper”, find_pair is purely geometric based (with user adjustable parameters) and allows for the identification of canonical Watson–Crick as well as non-canonical base pairs, made up of normal or modified bases, regardless of tautomeric or protonation state. For example, in the GpU paper”, we chose the following set of stringent parameters to ensure that the geometry of each identified base pair is nearly planar and supports at least one inter-base H-bond: (i) a vertical distance (stagger) between base planes ≤ 1.5 Å; (ii) an angle between base normal vectors ≤ 30°; and (iii) a pair of nitrogen and/or oxygen base atoms at a distance ≤ 3.3 Å. Other criteria (documented or otherwise), such as the distance between the origins of the two standard base reference frames, are just filters to speed up the calculations.
In a nutshell, find_pair has the following two core functionalities:
- The default is to generate input to the analysis routines in 3DNA (
analyze/cehs) for double helices. However, there are many more job to perform under the hood than just identifying base pairs: the base pairs must be in proper sequential order, and each strand must be in 5’ to 3’ direction, for the calculated step parameters (twist, roll etc) to make sense. Moreover, with the “-c” option, one gets an input file to Curves (but not Curves+, yet); with the “-s” or “-1” option, find_pair treats the whole structure as one single strand, and is useful for getting all backbone torsion angles.
- Detect all base pairs (regardless of double helical regions) and higher-oder (3+) base associations with the “-p” option. This feature (in its preliminary form) was there starting from at least v1.5, which was released at the end of 2002 (just before I left Rutgers), but it was intentionally undocumented. The source code of
find_pair (as part of 3DNA) was tested and shared within Rutgers (NDB and Dr. Olson’s laboratory) before any 3DNA paper was published, and served as the basis for several other projects. We also offered 3DNA (with source code) to a few RNA experts for comments; but we received either no responses or politely-worded negative ones. Things did not work out as (what I thought) they should have been, but that’s life and I have learned my lessons. The “-p” option was first explicitly mentioned in the 3DNA 2008 Nature Protocols paper, to illustrate how to identify the two pentaplets in the large ribosomal subunit of H. marismortui (1jj2).
It is interesting to mention the two papers I’ve recently come across: the first is on DNA-protein interactions and the second on RNA base-pairing, where new algorithms were developed to detect base pairs and their performances were compared with find_pair. In each of the two cases, it was claimed that find_pair missed certain pairs where the new methods succeeded. As it turned out, however, in the first case, simply relaxing find_pair’s default H-bond distance cut-off 4.0 Å to 4.5 Å, as used by the authors, virtually all the missing pairs were recovered. In the second case, the “-p” option, which should have been, was simply not specified.
After nearly a decade of extensive real-world applications and refinements, it is safe to say that find_pair is now a versatile and practical tool for nucleic acids structure analysis. Of course, I will continue to support and further refine find_pair as I see fit. Once in a while, I just cannot stop but to think that find_pair is to nucleic acids what DSSP is to proteins: simple and elegant. As more people become aware of its existence, I would expect find_pair to gain even more widespread usage, especially in RNA-structure related research areas.
While browsing Nucleic Acids Research recently, I noticed the paper titled Conformational analysis of nucleic acids revisited: Curves+ by Dr. Lavery et al. I read it through carefully during the weekend and played around with the software. Overall, I was fairly impressed, and also happy to see that “It [Curves+] adopts the generally accepted reference frame for nucleic acid bases and no longer shows any significant difference with analysis programs such as 3DNA for intra- or inter-base pair parameters.”
Anyone who has ever worked on nucleic acid structures (especially DNA) should be familiar with Curves, an analysis program that has been widely used over the past twenty years. Only in recent years has 3DNA become popular. By and large, though, it is my opinion that 3DNA and Curves are constructive competitors in nucleic acid structure analysis with complementary functionality. As I put it six years ago, before the 13th Conversation at Albany: “Curves has special features that 3DNA does not want to repeat/compete (e.g. global parameters, groove dimension parameters). Nevertheless, we provide an option in a 3DNA utility program (find_pair) to generate input to Curves directly from a PDB data file” on June 6, 2003, and emphasized again on June 09, 2003: “We also see Curves unique in defining global parameters, bending analysis and groove dimensions.” 3DNA’s real strength, as demonstrated in our 2008 Nature Protocols paper, lies in its integrated approach that combines nucleic acid structure analysis, rebuilding, and visualization into a single software package (see image below).

Now the nucleic acid structure community is blessed with the new Curves+, which “is algorithmically simpler and computationally much faster than the earlier Curves approach”, yet still provides its ‘hallmark’ curvilinear axis and “a full analysis of groove widths and depths”. When I read the text, I especially liked the INTRODUCTION section, which provides a nice summary of relevant background information on nucleic acid conformational analysis. An important feature of Curves+ is its integration of the analysis of molecular dynamics trajectories. In contrast, 3DNA lacks direct support in this area (even though I know of such applications from questions posted on the 3DNA forum), mostly due to the fact that I am not an ‘energetic’ person. Of special note is a policy-related advantage Curves+ has over 3DNA: Curves+ is distributed freely, and with source code available. On the other hand, due to Rutgers’ license constraints and various other (undocumented) reasons, 3DNA users are still having difficulty in accessing 3DNA v2.0 I compiled several months ago!
It is worth noting that the major differences in slide (+0.47 Å) and x-displacement (+0.77 Å) in Curves+ vs the old Curves (~0.5 Å and ~0.8 Å, respectively) are nearly exactly those uncovered a decade ago in Resolving the discrepancies among nucleic acid conformational analyses [Lu and Olson (1999), J. Mol. Biol., 285(4), 1563-75]:
Except for Curves, which defines the local frame in terms of the canonical B-DNA fiber structure (Leslie et al., 1980), the base origins are roughly coincident in the different schemes, but are significantly displaced (~0.8 Å along the positive x-axis) from the Curves reference. As illustrated below, this offset gives rise to systematic discrepancies of ~0.5 Å in slide and ~0.8 Å in global x-displacement in Curves compared with other programs, and also contributes to differences in rise at kinked steps. (p. 1566)
Please note that Curves+ has introduced new name list variables — most notably, lib= — and other subtle format changes, thus rendering the find_pair generated input files (with option ‘-c’) no longer valid. However, it would be easy to manually edit the input file to make it work for Curves+, since the most significant part — i.e., specifying paired nucleotides — does not change. Given time and upon user request, however, I would consider to write a new script to automate the process.
Overall, it is to the user community’s advantage to have both 3DNA and Curves+ or a choice between the two programs, and I am more than willing to build a bridge between them to make users’ lives easier.