 A video overview of DSSR
A video overview of DSSR
DSSR (Dissecting the Spatial Structure of RNA) is an integrated software tool for the analysis/annotation, model building, and schematic visualization of 3D nucleic acid structures (see the figures below and the video overview). It is built upon the well-known, tested, and trusted 3DNA suite of programs. DSSR has been made possible by the developer’s extensive user-support experience, detail-oriented software engineering skills, and expert domain knowledge accumulated over two decades. It streamlines tasks in RNA/DNA structural bioinformatics, and outperforms its ‘competitors’ by far in terms of functionality, usability, and support.
Wide citations. DSSR has been widely cited in scientific literature, including: (i) “Selective small-molecule inhibition of an RNA structural element” (Nature, 2015; Merck Research Laboratories), (ii) “The structure of the yeast mitochondrial ribosome” (Science, 2017), (iii) “RNA force field with accuracy comparable to state-of-the-art protein force fields” (PNAS, 2018; D. E. Shaw Research), (iv) “Predicting site-binding modes of ions and water to nucleic acids using molecular solvation theory” (JACS, 2019), (v) “RIC-seq for global in situ profiling of RNA-RNA spatial interactions” (Nature, 2020), and (vi) “DNA mismatches reveal conformational penalties in protein-DNA recognition” (Nature, 2020).
Broad integrations. To make DSSR as widely accessible as possible, I have initiated collaborations with the principal developers of Jmol and PyMOL. The DSSR-Jmol and DSSR-PyMOL integrations bring unparalleled search capabilities (e.g., ‘select junctions’ for all multi-branch loops) and innovative visualization styles into 3D nucleic acid structures. DSSR has also been adopted into numerous other structural bioinformatics resources, including: (i) URS, (ii) RiboSketch, (iii) RNApdbee, (iv) forgi, (v) RNAvista, (vi) VeriNA3d, (vii) RNAMake, (viii) ElTetrado, (ix) DNAproDB, (x) LocalSTAR3D, (xi) IPANEMAP, and (xii) RNANet.
Advanced features. DSSR may be licensed from Columbia University. DSSR Pro is the commercial version. It has more functionalities than DSSR basic (the free academic version), including: (i) homology modeling via in silico base mutations, a feature employed by Merck scientists, (ii) easy generation of regular helical models, including circular or super-helical DNA (see figures below), (iii) creation of customized structures with user-specified base sequences and rigid-body parameters, (iv) efficient processing of molecular dynamics (MD) trajectories, (v) detailed characterization of DNA-protein or RNA-protein spatial interactions, and (vi) template-based modeling of DNA-protein complexes (see figures below). DSSR Pro supersedes 3DNA. It integrates the disparate analysis and modeling programs of 3DNA under one umbrella, and offers new advanced features, through a convenient interface. For example, with the mutate module of DSSR Pro, one can automatically perform the following tasks: (i) mutate all bases to Us, (ii) mutate bases in hairpin loops to Gs, and (iii) mutate G–C Watson-Crick pairs to C–G, and A–U to U–A. Moreover, DSSR Pro includes an in-depth user manual and one-year technical support from the developer.
Quality control. DSSR is a solid software product that excels in RNA structural bioinformatics. It is written in strict ANSI C, as a single command-line program. It is self-contained, with zero runtime dependencies on third-party libraries. The binary executables for macOS, Linux, and Windows are just ~2MB. DSSR has been extensively tested using all nucleic-acid-containing structures in the PDB. It is also routinely checked with Valgrind to avoid memory leaks. DSSR requires no set up or configuration: it simply works.

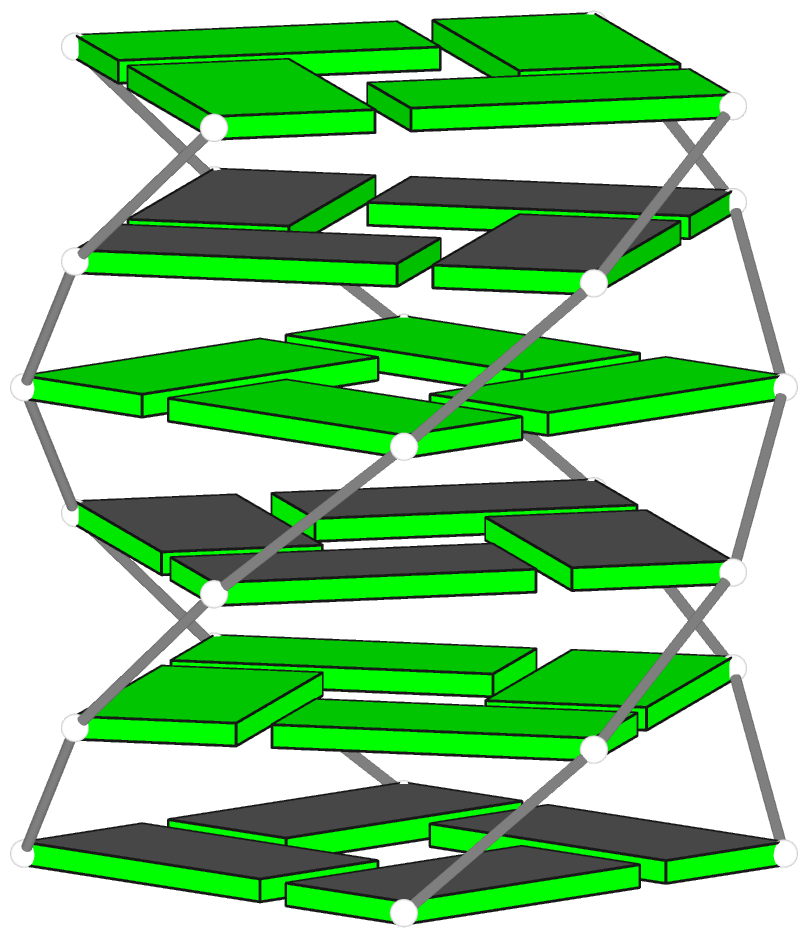
Theoretical models of G-quadruplexes, created using DSSR Pro.

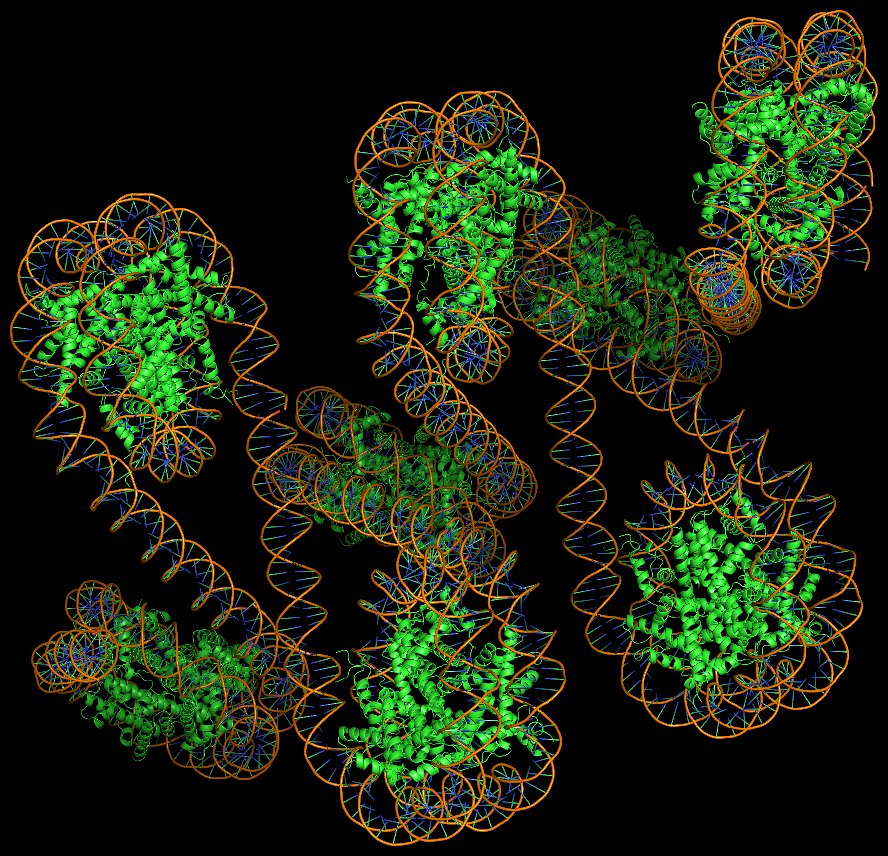
Template-based modeling of DNA-protein complexes using DSSR Pro.
Here are two chromatin-like models using PDB entry 4xzq as the template.


Circular DNA duplexes modeled using DSSR Pro.
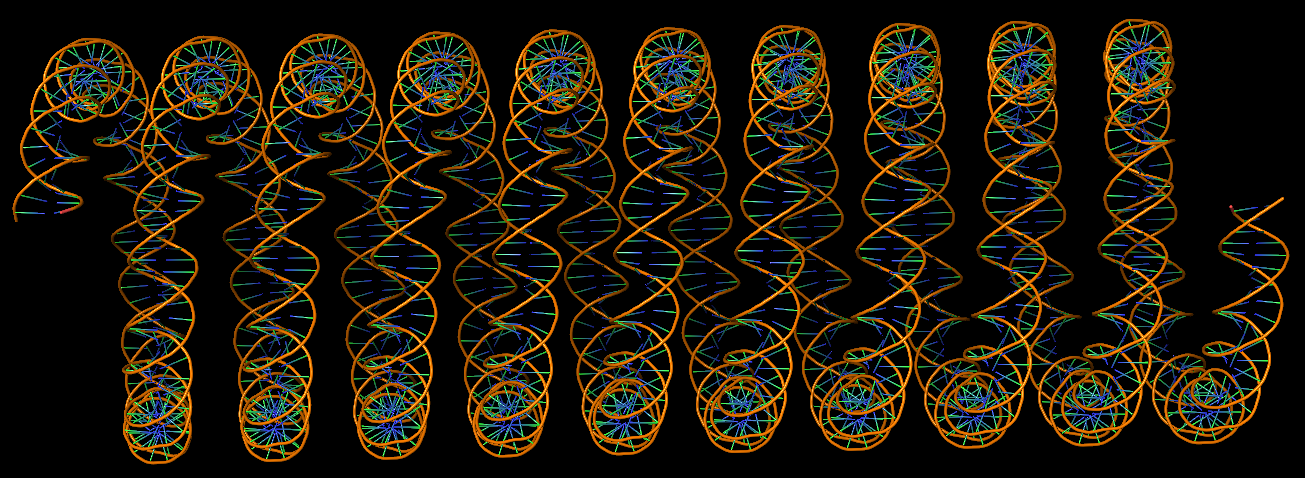
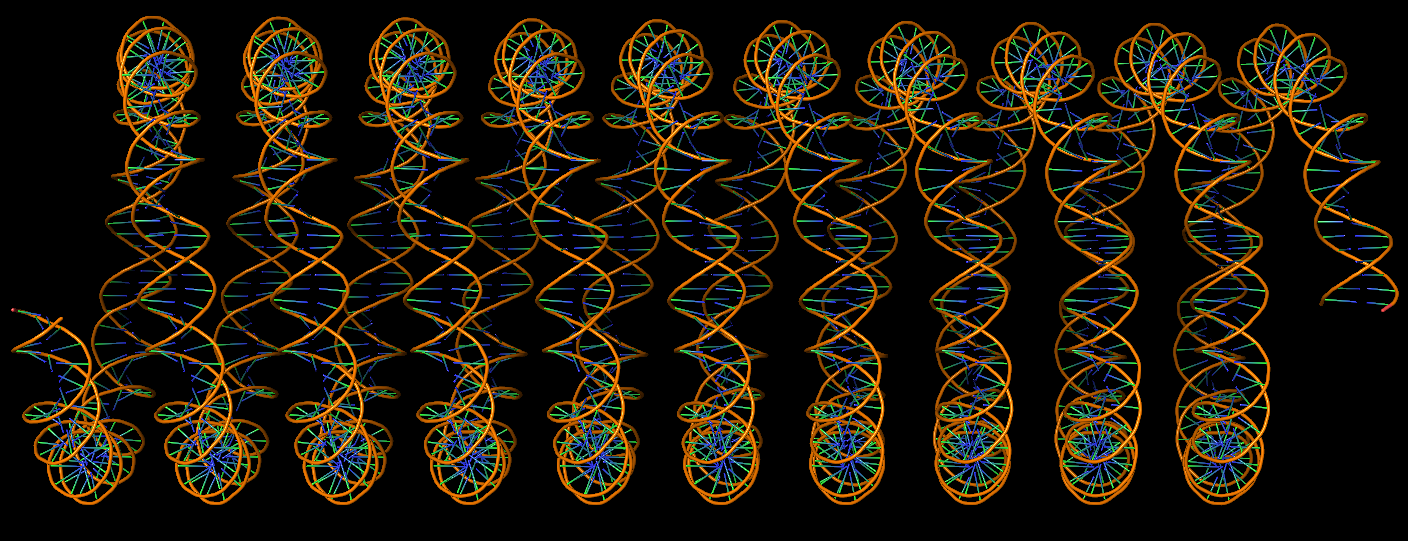
DNA super helices modeled using DSSR Pro.

Innovative cartoon-block schematics enabled by the DSSR-PyMOL integration for six representative PDB entries. Watson-Crick pairs are shown as long blocks with minor-groove edges in black (A, B), G-tetrads represented as square blocks and the metal ion as sphere ©, the ligand rendered as balls-and-sticks (D), and proteins depicted as purple cartoons (E, F). Color code for base blocks: A, red; C, yellow; G, green; T, blue; U, cyan; G-tetrad, green; WC-pairs, per base in the leading strand. Visit http://skmatic.x3dna.org.
Recommended in Faculty Opinions: “simple and effective”, “Good for Teaching”.
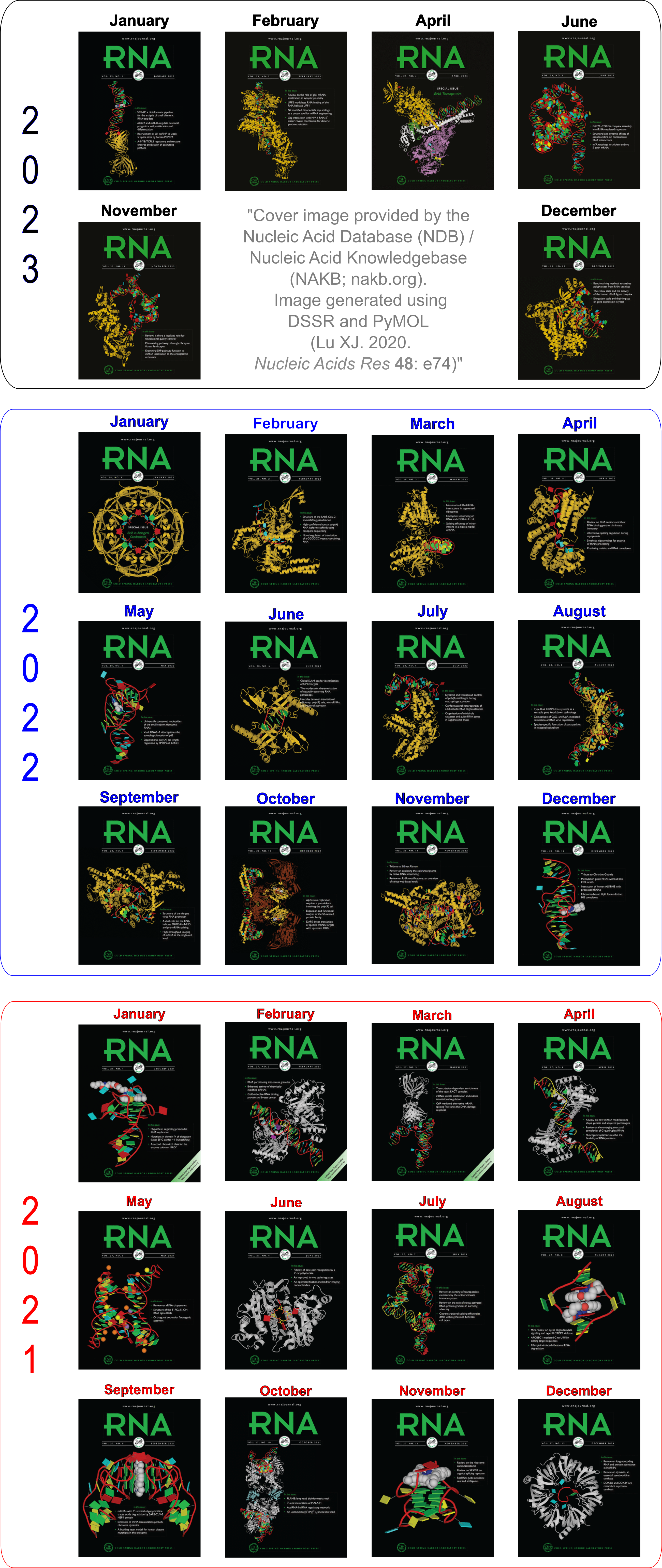
Employed by the NDB to create cover images of the RNA Journal.
The DSSR-PyMOL schematics have been featured in all 12 cover images (January to December) of the RNA Journal in 2021. Moreover, the January 2022 issue of RNA continues to highlight DSSR-enabled schematics (see the note below). In the current Covid-19 pandemic, this cover seems to be a fit for the upcoming Christmas holiday season.
Ebola virus matrix protein octameric ring (PDB id: 7K5L; Landeras-Bueno S, Wasserman H, Oliveira G, VanAernum ZL, Busch F, Salie ZL, Wysocki VH, Andersen K, Saphire EO. 2021. Cellular mRNA triggers structural transformation of Ebola virus matrix protein VP40 to its essential regulatory form. Cell Rep 35: 108986). The Ebola virus matrix protein (VP40) forms distinct structures linked to distinct functions in the virus life cycle. VP40 forms an octameric ring-shaped (D4 symmetry) assembly upon binding of RNA and is associated with transcriptional control. RNA backbone is displayed as a red ribbon; block bases use NDB colors: A—red, G—green, U—cyan; protein is displayed as a gold ribbon. Cover image provided by the Nucleic Acid Database (ndbserver.rutgers.edu). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).
Thanks to Dr. Cathy Lawson at the NDB for generating these cover images using DSSR and PyMOL for the RNA Journal. I’m gratified that the 2020 NAR paper is explicitly acknowledged: it’s the first time I’ve published as a single author in my scientific career.

Did you know that you can easily generate similar DSSR-PyMOL schematics via the http://skmatic.x3dna.org/ website? It is “simple and effective”, “good for teaching”, and has been highly recommended by Dr. Quentin Vicens (CU Denver) in FacultyOpinions.com.
The 12 PDB structures illustrated in the 2021 covers are:
- January 2021 “iMango-III fluorescent aptamer (PDB id: 6PQ7; Trachman III RJ, Stagno JR, Conrad C, Jones CP, Fischer P, Meents A, Wang YX, Ferre-D’Amare AR. 2019. Co-crystal structure of the iMango-III fluorescent RNA aptamer using an X-ray free-electron laser. Acta Cryst F 75: 547). Upon binding TO1-biotin, the iMango-III aptamer achieves the largest fluorescence enhancement reported for turn-on aptamers (over 5000-fold).”
- February 2021 “Human adenosine deaminase (E488Q mutant) acting on dsRNA (PDB id: 6VFF; Thuy-Boun AS, Thomas JM, Grajo HL, Palumbo CM, Park S, Nguyen LT, Fisher AJ, Beal PA. 2020. Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa532). Adenosine deaminase enzymes convert adenosine to inosine in duplex RNA, a modification that strongly affects RNA structure and function in multiple ways.”
- March 2021 “Hepatitis A virus IRES domain V in complex with Fab (PDB id: 6MWN; Koirala D, Shao Y, Koldobskaya Y, Fuller JR, Watkins AM, Shelke SA, Pilipenko EV, Das R, Rice PA, Piccirilli JA. 2019. A conserved RNA structural motif for organizing topology within picornaviral internal ribosome entry sites. Nat Commun 10: 3629).”
- April 2021 “Mouse endonuclease V in complex with 23mer RNA (PDB id: 6OZO; Wu J, Samara NL, Kuraoka I, Yang W. 2019. Evolution of inosine-specific endonuclease V from bacterial DNase to eukaryotic RNase. Mol Cell 76: 44). Endonuclease V cleaves the second phosphodiester bond 3′ to a deaminated adenosine (inosine). Although highly conserved, EndoV change substrate preference from DNA in bacteria to RNA in eukaryotes.”
- May 2021 “Manganese riboswitch from Xanthmonas oryzae (PDB id: 6N2V; Suddala KC, Price IR, Dandpat SS, Janeček M, Kührová P, Šponer J, Banáš P, Ke A, Walter NG. 2019. Local-to-global signal transduction at the core of a Mn2+ sensing riboswitch. Nat Commun 10: 4304). Bacterial manganese riboswitches control the expression of Mn2+ homeostasis genes. Using FRET, it was shown that an extended 4-way-junction samples transient docked states in the presence of Mg2+ but can only dock stably upon addition of submillimolar Mn2+.”
- June 2021 “Sulfolobus islandicus Csx1 RNase in complex with cyclic RNA activator (PDB id: 6R9R; Molina R, Stella S, Feng M, Sofos N, Jauniskis V, Pozdnyakova I, Lopez-Mendez B, She Q, Montoya G. 2019. Structure of Csx1-cOA4 complex reveals the basis of RNA decay in Type III-B CRISPR-Cas. Nat Commun 10: 4302). CRISPR-Cas multisubunit complexes cleave ssRNA and ssDNA, promoting the generation of cyclic oligoadenylate (cOA), which activates associated CRISPR-Cas RNases. The Csx1 RNase dimer is shown with cyclic (A4) RNA bound.”
- July 2021 “M. tuberculosis ileS T-box riboswitch in complex with tRNA (PDB id: 6UFG; Battaglia RA, Grigg JC, Ke A. 2019. Structural basis for tRNA decoding and aminoacylation sensing by T-box riboregulators. Nat Struct Mol Biol 26: 1106). T-box riboregulators are a class of cis-regulatory RNAs that govern the bacterial response to amino acid starvation by binding, decoding, and reading the aminoacylation status of specific transfer RNAs.”
- August 2021 “CAG repeats recognized by cyclic mismatch binding ligand (PDB id: 6QIV; Mukherjee S, Blaszczyk L, Rypniewski W, Falschlunger C, Micura R, Murata A, Dohno C, Nakatan K, Kiliszek A. 2019. Structural insights into synthetic ligands targeting A–A pairs in disease-related CAG RNA repeats. Nucleic Acids Res 47:10906). A large number of hereditary neurodegenerative human diseases are associated with abnormal expansion of repeated sequences. RNA containing CAG repeats can be recognized by synthetic cyclic mismatch-binding ligands such as the structure shown.”
- September 2021 “Corn aptamer complex with fluorophore Thioflavin T (PDB id: 6E81; Sjekloca L, Ferre-D’Amare AR. 2019. Binding between G quadruplexes at the homodimer interface of the Corn RNA aptamer strongly activates Thioflavin T fluorescence. Cell Chem Biol 26: 1159). The fluorescent compound Thioflavin T, widely used for the detection of amyloids, is bound at the dimer interface of the homodimeric G-quadruplex-containing RNA Corn aptamer.”
- October 2021 “Cas9 nuclease-sgRNA complex with anti-CRISPR protein inhibitor (PDB id: 6JE9; Sun W, Yang J, Cheng Z, Amrani N, Liu C, Wang K, Ibraheim R, Edraki A, Huang X, Wang M, et al. 2019. Structures of Neisseria meningitidis Cas9 complexes in catalytically poised and anti-CRISPR-inhibited states. Mol Cell 76: 938–952.e5). Nme1Cas9, a compact nuclease for in vivo genome editing. AcrIIC3 is an anti-CRISPR protein inhibitor.”
- November 2021 “Two-quartet RNA parallel G-quadruplex complexed with porphyrin (PDB id: 6JJI; Zhang Y, Omari KE, Duman R, Liu S, Haider S, Wagner A, Parkinson GN, Wei D. 2020. Native de novo structural determinations of non-canonical nucleic acid motifs by X-ray crystallography at long wavelengths. Nucleic Acids Res 48: 9886–9898).”
- December 2021 “Structure of S. pombe Lsm1–7 with RNA, polyuridine with 3’ guanosine (PDB id: 6PPV; Montemayor EJ, Virta JM, Hayes SM, Nomura Y, Brow DA, Butcher SE. 2020. Molecular basis for the distinct cellular functions of the Lsm1–7 and Lsm2–8 complexes. RNA 26: 1400–1413). Eukaryotes possess eight highly conserved Lsm (like Sm) proteins that assemble into circular, heteroheptameric complexes, bind RNA, and direct a diverse range of biological processes. Among the many essential functions of Lsm proteins, the cytoplasmic Lsm1–7 complex initiates mRNA decay, while the nuclear Lsm2–8 complex acts as a chaperone for U6 spliceosomal RNA.”

On December 9, 2021, at 15:00 CET, I will present a BioExcel webinar titled “X3DNA-DSSR, a resource for structural bioinformatics of nucleic acids.”
For the record, the screenshot of the announcement is shown below:

Today, I released a video overview of DSSR (http://docs.x3dna.org/dssr-overview/).
DSSR has a sizable user base. However, in my opinion, DSSR is still underutilized for what it has to offer. This overview video is intended not only to attract new DSSR users, but also to highlight features that even experienced users may overlook.
As documented in the Overview PDF, DSSR can be easily incorporated into other structural bioinformatics pipelines. Working with Robert Hanson and Thomas Holder respectively, I initiated the integrations of DSSR into Jmol and PyMOL, two of the most popular molecular viewers. The DSSR-Jmol and DSSR-PyMOL integrations lead to unparalleled search capabilities and innovative visualization styles of 3D nucleic acid structures. They also exemplify the critical roles that a domain-specific analysis engine may play in general-purpose molecular visualization tools.
On January 27, 2016, I wrote the blogpost Integrating DSSR into Jmol and PyMOL. Four years later, these integrations have led to two peer-reviewed articles, both published in Nucleic Acids Research (NAR). This blogpost (dated 2020-09-15) highlights key features in each case and reflects on my experience in these two exciting collaborations.
The DSSR-Jmol integration
Hanson RM and Lu XJ (2017). DSSR-enhanced visualization of nucleic acid structures in Jmol. The DSSR-Jmol integration excels in its SQL-like, flexible searching capability of structural features, as demonstrated at the website http://jmol.x3dna.org. This work fills a gap in RNA structural bioinformatics by enabling deep analyses and SQL-like queries of RNA structural characteristics, interactively. Here are some simple examples:
SELECT WITHIN(dssr, "nts WHERE is_modified = true") # modified nucleotides
SELECT pairs # all pairs
Select WITHIN(dssr, "pairs WHERE name = 'Hoogsteen'") # Hoogsteen pairs
SELECT WITHIN(dssr, "pairs WHERE name != 'WC'") # non-Watson-Crick pairs
SELECT junctions # all junctions loops
select within(dssr, "junctions WHERE num_stems = 4") # four-way junction loops
In a recently email communication, Bob wrote:
How are you doing? I’m smiling, because I am remembering our incredible, animated discussions and how fun it was to work together with you on Jmol and DSSR.
The DSSR-PyMOL integration
Lu XJ (2020). DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL. The DSSR-PyMOL integration brings unprecedented visual clarity to 3D nucleic acid structures, especially for G-quadruplexes. The four interfaces cover virtually all conceivable use cases. The easiest way to get started and quickly benefit from this work is via the web application at http://skmatic.x3dna.org.
I approached Thomas to write the DSSR-PyMOL manuscript together, in a similar way as the DSSR-Jmol paper. He wrote back, saying “I’m not interesting in being co-author of the paper”, adding:
But, if there is anything I can help you with, like revising the `dssr_block.py` script, or proof-reading the PyMOL related parts of the manuscript, I’ll be happy to do so.
Indeed, Thomas helped in several aspects of the DSSR-PyMOL project, as acknowledged in the paper:
I appreciate Thomas Holder (PyMOL Principal Developer, Schrödinger, Inc.) for writing the DSSR plugin for PyMOL, and for providing insightful comments on the manuscript and the web application interface.
Enhanced vs Innovative
Some viewers may noticed the difference in titles of the two NAR papers: “DSSR-enhanced visualization of nucleic acid structures in Jmol” vs. “DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL”. As a matter of fact, the initial title of the DSSR-PyMOL paper was DSSR-enhanced visualization of nucleic acid structures in PyMOL, as shown in the December 02, 2019 announcement post on the 3DNA Forum.
In an era where reproducibility of “scientific” publications has become an issue and “break-throughs” are often broken or hardly held, I hesitate to use phrases such as “innovative”, “novel”, “paradigm shift” etc. Instead, I often use the modest words “refinement”, “enhance”, “improved”, “revised” etc, and try to deliver more than claimed. However, reviewers may take solid work but modest writing as “incremental” or “unexciting”. Before submitting the DSSR-PyMOL paper, I changed the title to DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL. Does it mean that the DSSR-PyMOL integration is more innovative than the DSSR-Jmol case? Not necessarily. I do have a paper with “innovative” in its title.
Recently, while reading the Miskiewicz et al. review article How bioinformatics resources work with G4 RNAs, I noticed the term DSSR-G4DB under the category Databases with G4-related data. It refers to the website http://G4.x3dna.org (or g4.x3dna.org) that has been there since 2017 and weekly updated with new G-quadruplexes from the PDB. The DSSR-G4 resource, DSSR-Enabled Automatic Identification and Annotation of G-quadruplexes in the PDB, has already been cited several times in literature. However, I have not written up a paper on it yet, and thus have never thought carefully on a name for the resource. The term DSSR-G4DB sounds good to me, and I may well use it in the future.
Given below are the relevant quotations on DSSR and the DSSR-G4DB resource in the Miskiewicz et al. review article and my notes. The underlined headings (e.g., “Conclusion”) are those of the Miskiewicz et al. review article.
Methods: Databases with G4-related data
Currently, there exist 16 databases, which store information concerning quadruplexes. They fall into three categories: databases that collect primary or tertiary structures with experimentally verified G4s (DSSR-G4DB, G4IPDB, G4LDB, G4RNA, Lit392 and Lit638); databases storing data from high-throughput sequencing with mapped quadruplexes (GSE63874, GSE77282, GSE110582 and GSE129281); and databases of sequences with G4s identified in silico (Greglist, GRSDB2, G4-virus, Non-B DB v2.0, Plant-GQ and QuadBase2)
DSSR-G4DB [38] contains quadruplex nucleic acid structures found by DSSR in the Protein Data Bank [30], currently 354 entries. The data are annotated. Users can find information about G-tetrads, G4 helices and G4-stems and visualize the 3D models of G4 structures. Availability: webserver (http://g4.x3 dna.org). Recent update: 5 June 2020.
Note: DSSR-G4DB is updated weekly. The latest update is on 2020-09-09, with 362 G-quadruplexes auto-curated with DSSR from the PDB.
Methods: Tools that analyze and visualize 2D and 3D structure
Currently, four tools can analyze and visualize G4 structures. DSSR [38] … ElTetrado [31] … RNApdbee [66, 69] … 3D-NuS [65]
DSSR [38] processes the 3D structure of the RNA molecule and annotates its secondary structure. It is a part of the 3DNA suite [67] designed to work with the structures of nucleic acids. DSSR identifies, classifies and describes base pairs, multiplets and characteristic motifs of the secondary structure; helices, stems, hairpin loops, bulges, internal loops, junctions and others. It can also detect modules and tertiary structure patterns, includ- ing pseudoknots and kink-turns. The recent extension, DSSR- PyMOL [68], allows drawing cartoon-block schemes of the 3D structure and responds to the need for simplified visualization of quadruplexes. Input data formats: PDB, mmCIF and PDB ID. Availability: standalone program, web application (http://dssr.x3 dna.org/, http://skmatic.x3dna.org/).
Note: The other three tools all depend on or make use of DSSR and 3DNA:
- ElTetrado “ElTetrado depends on DSSR (Lu, Bussemaker and Olson, 2015) in terms of detection of base pairing and stacking.”
- RNApdbee uses 3DNA/DSSR as the default to identify base pairs.
- 3D-NuS employs 3DNA for structural analysis and model building.
“These filtrated structures (225 DNA and 166 RNA structures) have been used to derive the local base pair step and base pair parameters (Table S2 for DNA and Table S3 for RNA) using 3DNA software package [35] and are stored in the server for 3D-NuS modeling.”
“Soon after the user submits input for sequence-specific modeling, the server fetches the appropriate base pair step and base pair parameters from the database and creates a 3DNA style input file. Subsequently, the template model is built using the rebuild module of 3DNA software package and subjected to energy optimization using X-plor [56] to remove steric hindrance, specifically in the mismatch- containing duplexes (Fig. 1).”
Results: Computational experiments with structure-based tools
DSSR and ElTetrado identified quadruplexes in the input PDB files. Both programs focused on structural aspects of the input molecule, explicitly informing about quadruplexes and tetrads within the structure. DSSR provided an extensive analysis of 3D structures and output the data about G-tetrads, G-helices and G4-stems. It computed planarity for each G-tetrad and gave the sections area, rise and twist parameters for G4-helix and G4-stems. The program automatically assigned loop topologies according to the predefined types (P—parallel, D—diagonal and L—lateral) and their orientation (+/−). DSSR-PyMOL generated block schemes of both quadruplexes (Figure 4A3 and B3). ElTetrado also calculated planarity, rise and twist parameters and identified strand directions for both quadruplexes. It classified the quadruplexes and their component tetrads to ONZ classes. Finally, it generated the arc diagram (Figure 4A1 and B1) and two-line dot-bracket encoding of every quadruplex.
Note: DSSR contains an undocumented option --G4. With the ONZ variant, i.e., --g4=onz (case does not matter), DSSR also outputs the ONZ classification of G-tetrads from the same chain.
Conclusion
DSSR comprehensively examines the G4 structure, determines a variety of its parameters and provides the schematic 3D view.
It is worth noting that DSSR has been categorized under “Databases with G4-related data” and “Tools that analyze and visualize 2D and 3D structure” of the Methods section. It is not a tool that predicts G4 location in the sequence. There are 14 tools listed in “Table 2. Selected features of PQS prediction tools”, including G4Hunter and QGRS Mapper etc.
Recently, while visiting the NAR website on DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL, I noticed a big red circle ① near “View Metrics”. I was quite curious to see what it meant. After a few clicks, I was delighted to read the following recommendation in Faculty Opinions by Quentin Vicens:
I really enjoyed “playing” with the revised and expanded version of Dissecting the Spatial Structure of RNA (DSSR) described by Xiang-Jun Lu in this July issue of NAR. The software is known to generate ‘block view’ representations of nucleic acids that make many parameters more immediately visible, such as base composition, stacking, and groove depth. This new version includes Watson-Crick pairs shown as single rectangles, and G quadruplexes as large squares, making such regions more quickly distinguishable from other regions within an overall tertiary structure. I was amazed at how simple and effective the web interface was, and I liked the possibility to download a PyMOL session to look at molecules under different angles. If need be, blocks can be further edited in PyMOL using the provided plugin (see on page 35). I highly recommend it!
The DSSR-PyMOL schematics paper/website has been rated “Very Good”, and classified as “Good for Teaching”. See Vicens Q: Faculty Opinions Recommendation of [Lu XJ, Nucleic Acids Res 2020 48(13):e74]. In Faculty Opinions, 14 Aug 2020; 10.3410/f.738001682.793577327.
DSSR 2.0 is out. It integrates an unprecedented set of features into one computational tool, including analysis/annotation, schematic visualization, and model building of 3D nucleic acid structures. DSSR 2.0 supersedes 3DNA 2.4, which is still maintained but no additional features other than bug fixes are scheduled. See the DSSR 2.0 overview PDF.
DSSR delivers a great user experience by solving problems and saving time. Considering its usability, interoperability, features, and support, DSSR easily stands out among `competitors’. It exemplifies a `solid software product’. I strive to make DSSR a pragmatic tool that the structural bioinformatics community can count on.
DSSR 2.0 is licensed by Columbia University. The software remains free for academic users, with the basic user manual. The professional user manual (over 230 pages, including 7 appendices) is available for paid academic users or commercial users only. Licensing revenue helps ensure the long-term sustainability of the DSSR project.
Additionally, the paper “DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL” has recently been published in Nucleic Acids Research, 48(13):e74. Check the web interface.
The DSSR-PyMOL paper/website has been rated “very good” and classified as “Good for Teaching”. See Vicens Q: Faculty Opinions Recommendation of [Lu XJ, Nucleic Acids Res 2020 48(13):e74]. In Faculty Opinions, 14 Aug 2020; 10.3410/f.738001682.793577327
Recently I performed a survey of citations to thirteen 3DNA-related publications using Web of Science from Clarivate Analytics. The time range is from 2015 to 2020 (June 30), for a total of five-and-half years. The 1,050 citations span 223 scientific journals, covering a broad range of research fields such as biology, medicine, chemistry, physics, materials etc. Not surprisingly, the citing journals include Cell, Nature and sub-journals, Science, and PNAS.
Each of following six papers has been cited over 50 times, as detailed below. Adding the six numbers together, there are 962 citations, accounting for 92% of the total 1,050.
- [
402 times in 138 journals] Lu,X.-J. and Olson,W.K. (2003) 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res., 31, 5108–5121.
- [
201 times in 81 journals] Lu,X.-J. and Olson,W.K. (2008) 3DNA: A versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc., 3, 1213–1227.
- [
127 times in 71 journals] Zheng,G., Lu,X.J. and Olson,W.K. (2009) Web 3DNA––a web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res, 37, W240-6.
- [
115 times in 57 journals] Olson,W.K., Bansal,M., Burley,S.K., Dickerson,R.E., Gerstein,M., Harvey,S.C., Heinemann,U., Lu,X.-J., Neidle,S., Shakked,Z., Sklenar,H., Suzuki,M., Tung,C.-S., Westhof,E., Wolberger,C. and Berman,H.M. (2001) A standard reference frame for the description of nucleic acid base-pair geometry. J. Mol. Biol., 313, 229–237.
- [
66 times in 32 journals] Lu,X.-J., Bussemaker,H.J. and Olson,W.K. (2015) DSSR: An integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res., 43, e142.
- [
51 times in 41 journals] Lu,X.J., Shakked,Z. and Olson,W.K. (2000) A-form conformational motifs in ligand-bound DNA structures. J. Mol. Biol., 300, 819–40.
The top 21 journals that cite 3DNA papers 10 times or more are listed below. Nucleic Acids Research stands out, with a total of 148 citations, accounting for 14% of the total 1,050 citations.
148 Nucleic Acids Research
84 Journal of Physical Chemistry B
40 Physical Chemistry Chemical Physics
34 Biophysical Journal
33 Journal of Chemical Theory and Computation
29 Biochemistry
29 RNA
24 PLoS One
24 Scientific Reports
22 Journal of Biomolecular Structure & Dynamics
20 Bioinformatics
19 Journal of Chemical Information and Modeling
16 Nature Communications
15 Biopolymers
15 Journal of the American Chemical Society
12 Acta Crystallographica Section D: Structural Biology
12 Journal of Molecular Modeling
11 Chemistry: a European Journal
11 Journal of Chemical Physics
10 Journal of Biological Chemistry
10 Structure
As I am writing this blogpost on June 26, 2020, the registrations on the 3DNA Forum has reached 5,054. The numbers were 3,000 on October 15, 2016, 2,000 on on February 3, 2015, and 1,000 on February 27, 2013 respectively. For year 2020, the monthly registrations are 36 (January), 35 (February), 54 (March), 84 (April), 69 (May). As of June 26, the number is 56, which will more than likely pass 60 by the end of this month. The Covid-19 pandemic does not seem to having a negative effect on the registrations.
The over 5,000 registrations are from users all over the world. The 3DNA Forum remains spam free, and all questions are promptly answered. It is functioning well; certainly better than I originally imagined.
Overall, the Forum serves as a virtual platform for me to interact effectively with the ever-increasing user community. I greatly enjoy answering questions, fixing bugs, and making 3DNA/DSSR/SNAP better tools for real-world applications.