Announcing wDSSR: The Next-Generation Web Interface to X3DNA-DSSR
Dear 3DNA/DSSR Community,
We are thrilled to announce the official launch of wDSSR (https://web.x3dna-dssr.org/), the powerful new web interface to the X3DNA-DSSR analytical engine.
Developed by Drs. Shuxiang Li and Xiang-Jun Lu and supported by NIH grant R24GM153869, wDSSR represents a major leap forward from our highly popular 2019 Web 3DNA 2.0 framework. While Web 3DNA 2.0 has faithfully served the community for the analysis, visualization, and modeling of 3D nucleic acid structures, wDSSR was built from the ground up to take full advantage of modern web technologies and the latest DSSR backend capabilities.
A Modern, Streamlined Scientific Workflow
We have completely overhauled the user interface to provide a clean, intuitive, and task-driven experience. The core modeling and analysis tools are now seamlessly organized into a logical, single-word scientific workflow: Analyze, Rebuild, Model, Circularize, Mutate, Assemble, and Visualize.
Spotlight Feature: The "Assemble" Module
One of the most exciting upgrades is the newly renamed Assemble tab (formerly "Composite"). This advanced composite model builder allows you to effortlessly construct complex, higher-order models by linking any combination of nucleic acid duplexes or protein-DNA/RNA complexes. You can quickly connect up to six distinct target structures, ranging from simple linked A-DNA and B-DNA duplexes to large, protein-decorated structural assemblies.
Immediate Global Adoption
Although wDSSR has just launched, we are incredibly humbled to share that it is already seeing rapid worldwide adoption! According to recent network infrastructure data, the new interface is actively being used by researchers across North America, South America, Europe, and Asia. Within just a few days, we have recorded active sessions from prestigious institutions around the globe, including:
- The Weizmann Institute of Science in Israel
- Katholieke Universiteit Leuven in Belgium
- Queen's University in Canada
- Universidad Nacional Autonoma de Mexico (UNAM) in Mexico
- Emory University and the Wadsworth Centers Laboratories and Research in the United States
- Jawaharlal Nehru University and the China Education and Research Network in Asia
How to Cite
While a dedicated paper for wDSSR is currently in preparation, researchers should cite the server using its URL (https://web.x3dna-dssr.org/) alongside the 2019 Web 3DNA 2.0 paper and the foundational 2015 DSSR paper. Full details and funding acknowledgements can be found on our newly consolidated About page.
We invite you all to try out the new wDSSR platform! As always, your feedback is invaluable to us, and we encourage you to share your thoughts, questions, and structural models via the newly updated Questions & Feedback link in the wDSSR footer.
Happy modeling!
Recently, I came across the NAR breakthrough article titled 'Crystal Structure of the Class V GTP-Binding RNA Aptamer Bound to Its Ligand: GTP Recognition by a Topologically Complex Intermolecular G-Quadruplex' by Stafflinger et al. (2025). I read the paper carefully several times and really enjoyed both the content and the writing style. This work offers new insights into the structural complexities and ligand recognition capabilities of RNA. The 1.6-Å high-resolution X-ray crystal structure (PDB id: 9hrf) of the aptamer–GTP complex explains why the class V GTP aptamer has a particularly high affinity and specificity for GTP: The GTP ligand is integrated into one layer of a two-layered G-quadruplex, which is extended on one side by a UUGA tetrad and on the other side by a Watson–Crick base pair (C48–G52) that is stacked by an unpaired adenine (A49). Please see the figure below for further details.

DSSR can readily analyze this complex structure, by simply running the following command:
x3dna-dssr -i=9hrf.pdb --pair-water -o=9hrf.out
The --pair-water option enables the detection of water-mediated base pairs. For instance, it identifies the G41-water-A45 interaction, which is highlighted both below and within the UUGA tetrad shown in the lower left-corner of the figure above.
Base pairs
In total, DSSR identifies 37 base pairs, including:
- A7-G62 imino pair (
A-G Imino 08-VIII cWW cW-W)
- U9+U10 platform (
U+U Platform -- cSH cm+M)
- U9+G41 reverse wobble (
U+G rWobble 27-XXVII tWW tW+W)
- Pseudo-knotted U10-A45 interaction (
U-A WC 20-XX cWW cW-W) along with two G-tetrads forming G12+G46 and G13+G47 pairs (G+G -- 06-VI cHW cM+W)
- U29+G32 in the apical tetra-loop (
U+G -- -- tSW tm+W)
- Water-mediated G41-A45 pairing (
G-A Water -- cHH cM-M)
- Watson-Crick C48-G52 pair (
C-G WC 19-XIX cWW cW-W)
Note how DSSR's automatically derived pair names help orient readers to structural features, particularly stretches of Watson-Crick base pairs forming stems. DSSR categorizes base pairs using the widely accepted Saenger nomenclature and the Leontis-Westhof (LW) scheme. Moreover, it provides 3DNA/DSSR's unique M+N versus M–N distinction, which, along with a set of six parameters, allows for a rigorous characterization of base-pairing geometries.
DSSR also identifies isolated canonical base pairs that do not belong to any stem. In the PDB structure 9hrf, these are U10-A45 and C48-G52 (see below and in the upper-right corner of the figure above).
List of 37 base pairs
nt1 nt2 bp name Saenger LW DSSR
7 A.A7 A.G62 A-G Imino 08-VIII cWW cW-W
9 A.U9 A.U10 U+U Platform -- cSH cm+M
10 A.U9 A.G41 U+G rWobble 27-XXVII tWW tW+W
11 A.U10 A.A45 U-A WC 20-XX cWW cW-W
12 A.G12 A.G46 G+G -- 06-VI cHW cM+W
15 A.G13 A.G47 G+G -- 06-VI cHW cM+W
31 A.U29 A.G32 U+G -- -- tSW tm+W
33 A.G41 A.A45 G-A Water -- cHH cM-M
37 A.C48 A.G52 C-G WC 19-XIX cWW cW-W
Multiplets
DSSR identifies three multiplets, the first two of which are illustrated in the figure above.
List of 3 multiplets
1 nts=4 UUGA A.U9,A.U10,A.G41,A.A45
2 nts=4 GGGg A.G12,A.G42,A.G46,A.GTP100
3 nts=4 GGGG A.G13,A.G40,A.G43,A.G47
Stems, helices, and coaxial stacks
DSSR identifies three stems composed of (6, 8, 7) canonical pairs, each featuring continuous backbones. These three stems are coaxially arranged into a 23-pair-long helix through base-stacking interactions, irrespective of the type of base pair and the backbone connectivity (see below). The two additional base pairs within the 23-pair-long helix are the A7-G62 imino pair and the U29+G32 pair located in the apical tetra-loop. DSSR's geometric approach for identifying stems and helices aligns well with visual inspection, as demonstrated in the upper-left panel of the figure above.
Note: a helix is defined by base-stacking interactions, regardless of bp
type and backbone connectivity, and may contain more than one stem.
helix#number[stems-contained] bps=number-of-base-pairs in the helix
bp-type: '|' for a canonical WC/wobble pair, '.' otherwise
helix-form: classification of a dinucleotide step comprising the bp
above the given designation and the bp that follows it. Types
include 'A', 'B' or 'Z' for the common A-, B- and Z-form helices,
'.' for an unclassified step, and 'x' for a step without a
continuous backbone.
--------------------------------------------------------------------
helix#1[3] bps=23
strand-1 5'-GGGCGCAUAGGUCGGUCGCUGCU-3'
bp-type ||||||.|||||||||||||||.
strand-2 3'-CCCGUGGAUCCGGUCAGUGACGG-5'
helix-form AAA..AxAAA....xA..AAA.
1 A.G1 A.C68 G-C WC 19-XIX cWW cW-W
2 A.G2 A.C67 G-C WC 19-XIX cWW cW-W
3 A.G3 A.C66 G-C WC 19-XIX cWW cW-W
4 A.C4 A.G65 C-G WC 19-XIX cWW cW-W
5 A.G5 A.U64 G-U Wobble 28-XXVIII cWW cW-W
6 A.C6 A.G63 C-G WC 19-XIX cWW cW-W
7 A.A7 A.G62 A-G Imino 08-VIII cWW cW-W
8 A.U14 A.A60 U-A WC 20-XX cWW cW-W
9 A.A15 A.U59 A-U WC 20-XX cWW cW-W
10 A.G16 A.C58 G-C WC 19-XIX cWW cW-W
11 A.G17 A.C57 G-C WC 19-XIX cWW cW-W
12 A.U18 A.G56 U-G Wobble 28-XXVIII cWW cW-W
13 A.C19 A.G55 C-G WC 19-XIX cWW cW-W
14 A.G20 A.U54 G-U Wobble 28-XXVIII cWW cW-W
15 A.G21 A.C53 G-C WC 19-XIX cWW cW-W
16 A.U22 A.A39 U-A WC 20-XX cWW cW-W
17 A.C23 A.G38 C-G WC 19-XIX cWW cW-W
18 A.G24 A.U37 G-U Wobble 28-XXVIII cWW cW-W
19 A.C25 A.G36 C-G WC 19-XIX cWW cW-W
20 A.U26 A.A35 U-A WC 20-XX cWW cW-W
21 A.G27 A.C34 G-C WC 19-XIX cWW cW-W
22 A.C28 A.G33 C-G WC 19-XIX cWW cW-W
23 A.U29 A.G32 U+G -- -- tSW tm+W
List of 1 coaxial stack
1 Helix#1 contains 3 stems: [#1,#2,#3]
Loops
DSSR identifies two hairpin loops, one internal loop, and a [0,8,0] 3-way junction loop by default. As shown in the secondary structure depicted in the upper-left corner of the figure above, these results are evident. When excluding the two isolated canonical base pairs from consideration of secondary structures, DSSR identifies one hairpin loop composed of four nucleotides (U29 to G32), a bulge made up of thirteen nucleotides (G40 to G52), and an internal loop consisting of seven nucleotides on one strand (A7 to G13) and two nucleotides on the other strand (G61 and G62), precisely as described in the Stafflinger et al. (2025) paper.
The literature is inconsistent in its treatment of isolated canonical base pairs within RNA secondary structures. For instance, as detailed in the DSSR User Manual (Figure 3B), considering the isolated WC C−G pair (between C2658 and G2663) reveals the reported GUAA tetraloop (Correll et al., 2003) in PDB entry 1msy and a [5,4] asymmetric internal loop. Without this consideration, the tetraloop and internal loop delineated by the C−G pair merge, resulting in an enlarged hairpin loop spanning 17 nucleotides (from C2652 to G2668).
G-quadruplex
In the class V GTP aptamer-GTP complex structure (PDB entry: 9hrf), the two G-tetrads are formed by guanine nucleotides originating from two loop regions separated by an eight-base-pair A-form stem. This arrangement results in a complex and previously unobserved G-quadruplex topology. DSSR easily identifies the two G-tetrads that form a G4-helix (but not a G4-stem), which is defined by stacking interactions of G4-tetrads, regardless of backbone connectivity. In principle, a G4-helix may include more than one G4-stem via coaxial stacking interactions. The G4 helix/stem are defined in a similar manner to the double-stranded helix/stem as described above.
The relevant DSSR output is provided below. Observe the varying glycosidic bond patterns and groove dimensions, along with two non-G4-stem loops that include two terminal guanosines, which align with Figure 2B of Stafflinger et al. (2025).
List of 1 G4-helix
Note: a G4-helix is defined by stacking interactions of G4-tetrads, regardless
of backbone connectivity, and may contain more than one G4-stem.
helix#1[0] layers=2 INTRA-molecular
1 glyco-bond=---- sugar=---3 groove=---- WC-->Major nts=4 GgGG A.G12,A.GTP100,A.G42,A.G46
2 glyco-bond=-s-- sugar=-3-3 groove=wn-- WC-->Major nts=4 GGGG A.G13,A.G40,A.G43,A.G47
step#1 pm(>>,forward) area=7.84 rise=3.35 twist=32.6
strand#1 RNA glyco-bond=-- sugar=-- nts=2 GG A.G12,A.G13
strand#2 RNA glyco-bond=-s sugar=-3 nts=2 gG A.GTP100,A.G40
strand#3 RNA glyco-bond=-- sugar=-- nts=2 GG A.G42,A.G43
strand#4 RNA glyco-bond=-- sugar=33 nts=2 GG A.G46,A.G47
****************************************************************************
List of 2 non-stem G4 loops (INCLUDING the two terminal nts)
1 type=lateral helix=#1 nts=28 GUAGGUCGGUCGCUGCUUCGGCAGUGAG A.G13,A.U14,A.A15,A.G16,A.G17,A.U18,A.C19,A.G20,A.G21,A.U22,A.C23,A.G24,A.C25,A.U26,A.G27,A.C28,A.U29,A.U30,A.C31,A.G32,A.G33,A.C34,A.A35,A.G36,A.U37,A.G38,A.A39,A.G40
2 type=V-shaped helix=#1 nts=4 GGGG A.G40,A.G41,A.G42,A.G43
Pseudoknots
DSSR identifies one pseudoknot in the structure (PDB entry: 9hrf), enabled by the long-range U10-A45 WC pair (see the upper-right panel of the figure above). In literature, pseudoknots are defined by crossing WC pairs. However, in this structure, it is important to note the two synergistic G12+G46 and G13+G47 pairs within two layers of G-tetrads. The two loop regions are thus held tightly together through both pseudoknot and G-quadruplex formation. This observation suggests that the definition of pseudoknots may need to be expanded to include non-canonical pairs.
References
- Stafflinger H, Neißner K, Bartsch S, Pichler AK, Bartosik K, Dhamotharan K, et al. Crystal structure of the class V GTP-binding RNA aptamer bound to its ligand: GTP recognition by a topologically complex intermolecular G-quadruplex. Nucleic Acids Research. 2025;53:gkaf1315. https://doi.org/10.1093/nar/gkaf1315.
- Correll CC. The common and the distinctive features of the bulged-G motif based on a 1.04 A resolution RNA structure. Nucleic Acids Research. 2003;31:6806–18. https://doi.org/10.1093/nar/gkg908.

The latest release, DSSR v2.7.2-2026jan12, introduces the --pair-wise (or --pairwise) option, which combines the functionalities of the previous --pair-only and --non-pair options. Base-pair identification is a cornerstone of nucleic acid structural analysis, while non-pairing interactions like H-bonds and stacking are also vital structural features. However, the DSSR --non-pair feature is underutilized within the user community. By consolidating these into a single --pair-wise option, we streamline the process of identifying common interactions between nucleotides.
DSSR offers a wide range of nucleic acid structural features, but for users focusing on fundamental DNA/RNA analysis and annotation, the --pair-only option provides simplified functionality. This option instructs DSSR to generate only base-pairing information, which is essential for structural studies. When enabled, --pair-only significantly enhances performance, allowing DSSR to run approximately 10 times faster than in its default configuration. Running DSSR on the yeast phenylalanine tRNA (PDB 1ehz) with the --pair-only option leads to the following output instantaneously:
# x3dna-dssr -i=1ehz.pdb --pair-only
List of 34 base pairs
nt1 nt2 bp name Saenger LW DSSR
1 A.G1 A.C72 G-C WC 19-XIX cWW cW-W
2 A.C2 A.G71 C-G WC 19-XIX cWW cW-W
3 A.G3 A.C70 G-C WC 19-XIX cWW cW-W
4 A.G4 A.U69 G-U Wobble 28-XXVIII cWW cW-W
5 A.A5 A.U68 A-U WC 20-XX cWW cW-W
6 A.U6 A.A67 U-A WC 20-XX cWW cW-W
7 A.U7 A.A66 U-A WC 20-XX cWW cW-W
8 A.U8 A.A14 U-A rHoogsteen 24-XXIV tWH tW-M
9 A.U8 A.A21 U+A -- -- tSW tm+W
10 A.A9 A.A23 A+A -- 02-II tHH tM+M
11 A.2MG10 A.C25 g-C WC 19-XIX cWW cW-W
12 A.2MG10 A.G45 g+G -- -- cHS cM+m
13 A.C11 A.G24 C-G WC 19-XIX cWW cW-W
14 A.U12 A.A23 U-A WC 20-XX cWW cW-W
15 A.C13 A.G22 C-G WC 19-XIX cWW cW-W
16 A.G15 A.C48 G+C rWC 22-XXII tWW tW+W
17 A.H2U16 A.U59 u+U -- -- tSW tm+W
18 A.G18 A.PSU55 G+P -- -- tWS tW+m
19 A.G19 A.C56 G-C WC 19-XIX cWW cW-W
20 A.G22 A.7MG46 G-g -- 07-VII tHW tM-W
21 A.M2G26 A.A44 g-A Imino 08-VIII cWW cW-W
22 A.C27 A.G43 C-G WC 19-XIX cWW cW-W
23 A.C28 A.G42 C-G WC 19-XIX cWW cW-W
24 A.A29 A.U41 A-U WC 20-XX cWW cW-W
25 A.G30 A.5MC40 G-c WC 19-XIX cWW cW-W
26 A.A31 A.PSU39 A-P -- -- cWW cW-W
27 A.OMC32 A.A38 c-A -- -- c.W c.-W
28 A.U33 A.A36 U-A -- -- tSH tm-M
29 A.5MC49 A.G65 c-G WC 19-XIX cWW cW-W
30 A.U50 A.A64 U-A WC 20-XX cWW cW-W
31 A.G51 A.C63 G-C WC 19-XIX cWW cW-W
32 A.U52 A.A62 U-A WC 20-XX cWW cW-W
33 A.G53 A.C61 G-C WC 19-XIX cWW cW-W
34 A.5MU54 A.1MA58 t-a rHoogsteen 24-XXIV tWH tW-M
With the --non-pair option, DSSR identifies H-bonding and base-stacking interactions between two nucleotides that do not form a pair. This option is an additional feature integrated into DSSR, expanding its capabilities by including these non-pairing interactions in the main output alongside pairing information, among other functionalities. Running DSSR on the yeast phenylalanine tRNA (PDB 1ehz) with the --non-pair option identifies 91 non-pairing interactions, with the first 16 listed below.
# x3dna-dssr -i=1ehz.pdb
List of 91 non-pairing interactions
1 A.G1 A.C2 stacking: 5.4(2.6)--pm(>>,forward) interBase-angle=5 connected min-baseDist=3.26
2 A.G1 A.A73 stacking: 2.4(1.2)--mm(<>,outward) interBase-angle=3 min-baseDist=3.17
3 A.C2 A.G3 stacking: 0.5(0.0)--pm(>>,forward) interBase-angle=9 connected min-baseDist=3.41
4 A.G3 A.G4 stacking: 3.2(1.8)--pm(>>,forward) interBase-angle=10 H-bonds[1]: "O2'(hydroxyl)-O4'[3.11]" connected min-baseDist=3.24
5 A.G3 A.G71 stacking: 2.6(0.3)--mm(<>,outward) interBase-angle=5 min-baseDist=3.02
6 A.G4 A.A5 stacking: 5.6(3.5)--pm(>>,forward) interBase-angle=6 connected min-baseDist=3.13
7 A.A5 A.U6 stacking: 5.9(4.3)--pm(>>,forward) interBase-angle=9 connected min-baseDist=3.12
8 A.U6 A.U7 stacking: 0.6(0.0)--pm(>>,forward) interBase-angle=20 connected min-baseDist=3.11
9 A.U7 A.5MC49 stacking: 1.2(0.0)--pm(>>,forward) interBase-angle=7 H-bonds[1]: "O2'(hydroxyl)-OP2[2.68]" min-baseDist=3.64
10 A.U8 A.C13 stacking: 2.0(0.0)--pp(><,inward) interBase-angle=13 min-baseDist=3.34
11 A.U8 A.G15 stacking: 0.5(0.0)--mm(<>,outward) interBase-angle=14 min-baseDist=3.27
12 A.A9 A.C11 interBase-angle=27 H-bonds[1]: "O2'(hydroxyl)-N4(amino)[2.90]" min-baseDist=3.72
13 A.A9 A.C13 interBase-angle=9 H-bonds[1]: "OP2-N4(amino)[3.01]" min-baseDist=4.65
14 A.A9 A.G22 stacking: 0.1(0.0)--mp(<<,backward) interBase-angle=13 min-baseDist=3.37
15 A.A9 A.G45 stacking: 1.6(0.5)--pp(><,inward) interBase-angle=10 min-baseDist=3.30
16 A.A9 A.7MG46 stacking: 1.6(0.7)--mm(<>,outward) interBase-angle=4 H-bonds[1]: "O5'-N2(amino)[3.34]" min-baseDist=3.38
......
DSSR calculates base-stacking by determining the overlap area (in Ų) between two interacting bases. The calculation involves projecting the atoms of the two bases onto their mean plane to define the overlapping region, from which the area is derived. In the output, values in parentheses represent the overlap area based solely on ring atoms, while those outside parentheses include contributions from exocyclic atoms as well (see Lu and Olson, 2003; Lu et al., 2015).
Base-stacking interactions are classified into one of four categories:
- pm (>>, forward): Interaction occurs on the plus-minus faces of the two bases in a forward direction.
- mp (<<, backward): Interaction occurs on the minus-plus faces of the two bases in a backward direction.
- mm (<>, outward): Interaction occurs between two minus faces oriented outward.
- pp (><, inward): Interaction occurs between two plus faces oriented inward.
In this classification:
p represents the plus face of the base ring, andm represents the minus face.
These categories are defined by the direction of the z-axis in the standard base reference frame (Olson et al., 2001). The symbols (>>, <<, <>, and ><) follow Parisien et al. (2009), with the exception that:
- pm (>>) is referred to as "forward" instead of "upward," and
- mp (<<) is referred to as "backward" instead of "downward."
The new --pair-wise option functions similarly to the --pair-only option by generating a separate output file. However, unlike --pair-only, it also includes non-pairing interactions in this file. DSSR runs faster than the full analysis because it characterizes only base-pairing and non-pairing interactions. Additionally, the --more and --json options are supported, enabling users to derive more detailed features (e.g., local base-pair parameters and H-bonds in base pairs) and easily parse them using JSON output.
Running DSSR on the yeast phenylalanine tRNA (PDB 1ehz) with the --pair-wise option identifies 34 base pairs and 91 non-pairing interactions, as expected. When combined with the --more and --json options, the output is summarized below.
# x3dna-dssr -i=1ehz.pdb --pair-wise --more --json | fx
{
"num_pairs": 34,
"pairs": […],
"num_nonPairs": 91,
"nonPairs": […],
"program": "DSSR v2.7.2-2026jan12 by xiangjun@x3dna.org"
}
Please refer to the DSSR User Manual for comprehensive explanations of all available features.
References
- Lu X-J, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–21. https://doi.org/10.1093/nar/gkg680.
- Lu X-J, Bussemaker HJ, Olson WK. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 2015;:gkv716. https://doi.org/10.1093/nar/gkv716.
- Olson WK, Bansal M, Burley SK, Dickerson RE, Gerstein M, Harvey SC, et al. A standard reference frame for the description of nucleic acid base-pair geometry. Journal of Molecular Biology. 2001;313:229–37. https://doi.org/10.1006/jmbi.2001.4987.
- Parisien M, Cruz JA, Westhof É, Major F. New metrics for comparing and assessing discrepancies between RNA 3D structures and models. RNA. 2009;15:1875–85. https://doi.org/10.1261/rna.1700409.
 March 2026
March 2026
 February 2026
February 2026
Cover image provided by X3DNA-DSSR, an NIGMS National Resource for Structural Bioinformatics of Nucleic Acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).
As the developer of DSSR, I am thrilled to see its application in cutting-edge research across multiple disciplines. Below is a list of four recent publications that highlight how DSSR has been utilized, underscoring its versatility and significance in structural bioinformatics.
In the Geng et al. (2025) Nucleic Acids Research (NAR) paper, titled 'Revealing hidden protonated conformational states in RNA dynamic ensembles', DSSR is simply cited as follows:
All bp geometries, hydrogen-bond, backbone, stacking, and sugar dihedral angles were calculated using X3DNA-DSSR [77].
In the preprint by Gordan et al. (2025), titled 'High-throughput characterization of transcription factors that modulate UV damage formation and repair at single-nucleotide resolution', DSSR is cited as follows:
Step base stacking, base pair shift, base pair slide, interbase angle, pseudorotation angle, and sugar puckering classifications of nucleobases were computed using X3DNA-DSSR (v2.5.0)75. Base stacking was defined as the overlapping polygon area in Å2 when projecting the dipyrimidine base ring atoms (excluding exocyclic atoms) into the mean base pair plane76. The sugar ring pseudorotation phase angle of each pyrimidine was also calculated using X3DNA-DSSR as described by Altona, C. & Sundaralingam, M.77 Interbase angle was defined as sqrt(propeller2+buckle2) per the X3DNA-DSSR documentation.
Figure 6: TF Binding Induces Structural Distortion Favorable to UV Dimerization is highly informative, particularly panel (a), which illustrates the ensemble of structural parameters that predispose dipyrimidines to cyclobutane pyrimidine dimers (CPD) or 6-4 pyrimidine-pyrimidones (6-4 PP) formation. DSSR is designed as an integrated software tool, offering a comprehensive suite of structural parameters not found in any other single tool I am aware of. Despite this, the innovative use of DSSR by Gordan et al. exceeds my expectations and demonstrates its versatility.
In the preprint by Kubaney et al. (2025) from the Baker group, titled 'RNA sequence design and protein-DNA specificity prediction with NA-MPNN', DSSR is cited as follows:
On the pseudoknot subset, we evaluate additional structure‐ and reactivity‐based metrics. DSSR v2.3.241 is used to extract the ground‐truth secondary structure from the native crystal structures. For each designed sequence, RibonanzaNet predicts 2A3 reactivity profiles, from which we compute predicted OpenKnot scores (see https://github.com/eternagame/OpenKnotScore)31 using the predicted reactivity together with the DSSR ground truth.
In a recent NSMB paper from the Baker group, titled 'Computational design of sequence-specific DNA-binding proteins', 3DNA is cited as follows:
RIF docking of scaffolds onto DNA targets (DBP design step 1) Structures of B-DNA for each target (Supplementary Table 2) were generated by (1) using the DNA portion of PDB 1BC8 (ref. 60), PDB 1YO5 (ref. 61), PDB 1L3L (ref. 51) or PDB 2O4A (ref. 62) or (2) using the software X3DNA63, followed by a constrained Rosetta relax of the DNA structure.
Please note that 3DNA has been replaced by DSSR. The functionality for constructing B-DNA models, previously provided by 3DNA, is now directly available in DSSR via its fiber and rebuild modules.
In the preprint by Si et al. (2025), titled 'End-to-End Single-Stranded DNA Sequence Design with All-Atom Structure Reconstruction', DSSR is cited as follows:
Since ViennaRNA and NUPACK require secondary structures as input, we used DSSR35 to extract secondary structures from the corresponding ssDNA three-dimensional structures.
The above use cases are merely a sample of how DSSR is utilized in the scientific literature. It is reasonable to state that DSSR has emerged as a de facto standard tool within the field of nucleic acid structural bioinformatics. Overall, DSSR is a mature, robust, and efficient software product that is actively developed and maintained. I am committed to making DSSR synonymous with quality and value. Its unmatched functionality, usability, and support save users significant time and effort compared to alternative solutions.
DSSR is available free of charge for academic users. Additionally, it has been integrated into other high-profile bioinformatics resources, including NAKB, PDB-redo, and N•ESPript.
References
- Geng A, Roy R, Ganser L, Li L, Al-Hashimi HM. Revealing hidden protonated conformational states in RNA dynamic ensembles. Nucleic Acids Research. 2025;53:gkaf1366. https://doi.org/10.1093/nar/gkaf1366.
- Gordan R, Wasserman H, Chi B, Bohm K, Duan M, Sahay H, et al. High-throughput characterization of transcription factors that modulate UV damage formation and repair at single-nucleotide resolution. 2025. https://doi.org/10.21203/rs.3.rs-8197218/v1.
- Kubaney A, Favor A, McHugh L, Mitra R, Pecoraro R, Dauparas J, et al. RNA sequence design and protein–DNA specificity prediction with NA-MPNN. 2025. https://doi.org/10.1101/2025.10.03.679414.
- Glasscock CJ, Pecoraro RJ, McHugh R, Doyle LA, Chen W, Boivin O, et al. Computational design of sequence-specific DNA-binding proteins. Nat Struct Mol Biol. 2025;32:2252–61. https://doi.org/10.1038/s41594-025-01669-4.
- Si Y, Xu Y, Chen L. End-to-end single-stranded DNA sequence design with all-atom structure reconstruction. 2025. https://doi.org/10.64898/2025.12.05.692525.
By following DSSR citations, I recently came across the paper by Saon et al. (2025), titled 'Identification and characterization of shifted G•U wobble pairs resulting from alternative protonation of RNA.' This paper provides a detailed analysis of shifted G-U wobble pairs in RNA, characterized by the opposite positioning of G vs. U in the standard G-U wobble pair (see figure below). Conventionally, a G-U wobble has the U located in the major groove, whereas a shifted G-U wobble has the G located in the major groove.
Specifically, the shifted G-U wobble pair involves an H-bond between the N2(G) and N3(U) atoms, which would be donor-donor if U were in its neutral form. There are three ways to rationalize the formation of this H-bond: (1) anionic U as originally proposed by Westhof et al. (2023), (2) U-enolate, and (3) G-imino tautomeric forms as illustrated by Saon et al. (2025). Since the position of the H-atoms cannot be determined from X-ray diffraction and cryo-EM structures, it is not possible (in my understanding) to determine which of these three mechanisms is correct—perhaps it involves a combination of them. What is clear is that the shifted G-U wobble pair is supported by strong experimental evidence from diverse sources. The authors identified 373 high-confidence shifted G-U wobble pairs across four separate structural clusters, spanning all three domains of life.
 Structure of standard and shifted G-U wobble pairs. The examples are taken from PDB entry 8B0X (Fromm et al., 2023) and generated using DSSR and PyMOL. Atom names in the Watson-Crick edges are shown in red and blue for oxygen and nitrogen, respectively. Hydrogen bonds are depicted as dashed lines in magenta. The unusual N2(G)...N3(U) hydrogen bond is marked with a star; it would be donor-donor if U were in its neutral form. The shaded illustration at the bottom is taken from Saon et al., showing shifted G-U wobble pairs in anionic, U-enolate, and G-imino tautomeric forms.
Structure of standard and shifted G-U wobble pairs. The examples are taken from PDB entry 8B0X (Fromm et al., 2023) and generated using DSSR and PyMOL. Atom names in the Watson-Crick edges are shown in red and blue for oxygen and nitrogen, respectively. Hydrogen bonds are depicted as dashed lines in magenta. The unusual N2(G)...N3(U) hydrogen bond is marked with a star; it would be donor-donor if U were in its neutral form. The shaded illustration at the bottom is taken from Saon et al., showing shifted G-U wobble pairs in anionic, U-enolate, and G-imino tautomeric forms.
I'm glad to see that DSSR has been used in the analysis, as shown in the following excerpts from the paper.
The selected structures were then characterized by Dissecting the Spatial Structure of RNA (DSSR) software [34]. This step output base pair, hydrogen bond, stacking, glycosidic angle, and sugar pucker information for each structure file.
From the DSSR base pair information, all G•U base pairs were identified and filtered as wobble or non-wobble base pairs. All base pairs called by DSSR as G•U wobbles were considered for the next steps of the analysis as standard wobbles. Any base pairs containing hydrogen bonds between G(N1) and U(O4), as well as G(N2) and U(N3) (see Fig. 1) were binned to shifted wobble base pairs.
From the base pair information extracted from the DSSR characterization output, the non-redundant G•U wobbles were binned based on their location in one of the five secondary structure motifs: (1) inside stem, with one WCF base pair above and one below, (2) terminal, with at least one WCF base pair above, (3) terminal, with at least one WCF base pair below, (4) unstructured, where no WCF base pair is right above or below and the wobble does not occur at the closing base pair of a hairpin loop with a maximum of 10 nucleotides, and (5) inside a loop.
Next, for each of the five members, we retrieved the 3D structure of the 20 residues from the respective pdb files and obtained the underlying secondary structures for each of the five files in dot bracket notation using DSSR [34].
DSSR implements a geometric approach to identify hydrogen bonds, including unconventional donor/acceptor combinations (e.g., the N3-to-N3 hydrogen bond in the hemiprotonated cytosine–cytosine base pair in the i-motif). It is capable of identifying all pairs that actually exist in a given structure, whether they are canonical (Watson-Crick or G-U wobble) or non-canonical. The latter pairs may include normal or modified nucleotides, regardless of their tautomeric or protonation state.
Thus, DSSR detects standard G-U wobble pairs and names them as such ('Wobble'). Moreover, it also detects shifted G-U wobble pairs and previously named them as '~Wobble,' meaning similar to a standard wobble pair. Note that the '~Wobble' designation is based on the geometric approach of DSSR, which involves the cW-W relative orientation of the two bases and a large shear value. It is not limited to wobble pairs between G and U.
After reading the Saon et al. paper, I have revised DSSR to specifically characterize shifted G-U wobble pairs and named them as 'sWobble.' The term 'shifted-Wobble' would be too long for the DSSR text output, and using 's' also reflects the shear parameter, which is key in characterizing wobble pairs. As a concrete example, the following DSSR command
x3dna-dssr -i=8B0X.cif --pair-only --more -o=8B0X-pairs.out
would generate the below output in the file 8B0X-pairs.out. Note the name sWobble, the hydrogen bond N3(imino)*N2(amino)[3.26] with a * to indicate an unusual donor/acceptor combination, and the -2.33 shear value.
607 A.U1086 A.G1099 U-G sWobble -- cWW cW-W
[-171.2(anti) ~C3'-endo lambda=33.9] [-170.0(anti) ~C3'-endo lambda=59.2]
d(C1'-C1')=11.57 d(N1-N9)=9.60 d(C6-C8)=10.07 tor(C1'-N1-N9-C1')=8.8
H-bonds[2]: "N3(imino)*N2(amino)[3.26],O4(carbonyl)-N1(imino)[2.64]"
interBase-angle=26 Simple-bpParams: Shear=-2.23 Stretch=0.69 Buckle=22.1 Propeller=-13.8
bp-pars: [-2.33 0.13 -0.80 24.83 -7.98 -20.38]
The new DSSR version can automatically detect all 373 high-confidence shifted G-U wobble pairs listed in Table S3 of the Saon et al. paper. It will be released soon. This is yet another example of how DSSR is being actively improved to better serve the research community.
References
- Saon,M.S. et al. (2025) Identification and characterization of shifted G•U wobble pairs resulting from alternative protonation of RNA. Nucleic Acids Research, 53, gkaf575.
- Westhof,E. et al. (2023) Anionic G•U pairs in bacterial ribosomal rRNAs. RNA, 29, 1069–1076.
- Fromm,S.A. et al. (2023) The translating bacterial ribosome at 1.55 Å resolution generated by cryo-EM imaging services. Nat Commun, 14, 1095.
By following DSSR citations, I recently noticed a bioRxiv preprint, titled "Assessment of nucleic acid structure prediction in CASP16" by Kretsch et al. The portion where DSSR is mentioned is as follows:
Secondary structures were extracted from CASP16 models with DSSR (v1.9.9-2020feb06). Some models, in particular due to large clashes, failed to run (Supplemental Table 1). The base-pair list was extracted from the table in the output file directly because the dot-bracket structure produced by DSSR, in particular for multimers, can contain errors.
While pleased to see DSSR cited in this significant study, I am concerned about the reported issues and would like to investigate the specific structures and error messages encountered. To better understand the problems and potentially find solutions, I have reached out to the authors for further details. Here is the message I sent initially:
You said DSSR failed to run on some models with large clashes. Could you please share the specific models and the error messages you encountered? I would also be interested in seeing the exact errors you observed in the DSSR-derived DBN for multi-mers. It would be a great opportunity for me to improve DSSR in this area, which would benefit both your group and the broader community. If you are willing to share them, please provide details—preferably on the **public 3DNA Forum**. Don’t hesitate to share openly any bugs or limitations you’ve encountered with DSSR.
The authors responded promptly and provided detailed information about the specific models and error messages encountered. After several
iterations, I successfully resolved the issues and released an updated version of DSSR, namely v2.5.4-2025jun04. You can find the release notes
here. This experience underscores the importance of proactively engaging with the community to enhance the functionality and reliability of a software tool.
In this blog post, I aim to share the specifics of these issues and the steps taken to address them. For ease of reading, I have formatted the response/feedback from the authors in red block quotes, and my enquiries/comments in blue. The beginning round of correspondence is as below.
Do note, the predictors in casp submit some truly atrocious models --- eg 14 atoms all at the exact same x-y-z coordinate. These errors would be with his v1.9.9-2020feb06 install though not your latest version. Would you still like them?
Yes, I would like to see how DSSR behaves with these models. Ideally, it should not crash, but output some warning messages. Only through such testing can we improve the robustness of DSSR. Overall, the more feedback I get, the better.
Buffer overflow bug in DSSR
Most of errors I had with dssr were due to clashes and all zero xyz predictions by predictors, for all of which dssr did not give an error message when dssr failed. There was a case where the prediction looked reasonable but dssr failed with the error message `dssr error*** buffer overflow detected ***`. Please see attached for the 2 pdbs that gave this error.
The two PDB files I received were R1283v3TS294_1o and R1283v3TS294_2o, as listed in Supplementary Table 1: "List of unscored models," with the "Reasons" column indicating a dssr error*** buffer overflow detected ***. I immediately acknowledged receipt of these files, as shown in the following message:
Thank you for sending me the two PDB files which caused DSSR to fail. I can verify the issue and will try to fix the bug ASAP. I'll keep you posted.
Using these data files, I was able to quickly fix the buffer overflow bug. The following is my response to the authors within one day after receiving the files:
With your sample PDB files, I have traced the issue that caused DSSR to fail. The bug was due to a 53-way (`R1283v3TS294_1o`) and 40-way (`R1283v3TS294_2o`) junction loops which are far from the norm. DSSR sets a default limit for the summary line for each loop which is more than sufficient for all normal PDB entries, but falls short for these unusual cases, leading to out of array boundaries. See the attached DSSR output after the bug fix for more details.
This is a clear example where user feedback is crucial for improving the software, which makes it better serve the community.
Zero xyz coordinates and large clashes
After fixing the out-of-bound bug, I also requested other problematic predicted models from the authors, as shown in the following message:
Along the line, please provide the sample PDB files:
- with zero xyz predictions -- I am curious to see what it looks like.
- where the DSSR-derived DBN is problematic for multi-mers
After solving these issues, I will release a new version of DSSR that would make your analysis more straightforward, and benefit other users as well.
The authors responded with the following message:
Thanks for looking into this. Here are some more examples with superimposed structures, large clash, and all zero xyzs in the zip file.
The ZIP file (error_examples.zip) contains three folders (all_zero_xyz, clash and superimposed), each with some problematic models in PDB format. Once again, I promptly acknowledged receipt of the files and was able to reproduce the reported issues.
Garbage in, garbage out. Given these problematic models, one should not expect DSSR to extract any meaningful information from them. Nonetheless, I am committed to enhancing the software so that it can handle such cases more effectively by providing clear error messages and terminating gracefully rather than crashing.
After several days of thinking, elaboration, intensive coding, and testing, I solved the problems. I then communicated the results to the authors in the following detailed message:
Thanks for the sample PDB files (`error_examples`) with all zero XYZ coordinates, large clashes, and superimposed structures. They helped me to understand the issues, think in context, and find solutions. Let's look them one by one:
1. `all_zero_xyz`: These two files `R1211TS159_1` and `R1211TS159_2` have identical contents, except for the MODEL IDs (1 and 2, respectively). Atoms with all-zero XYZ coordinates are a special case of duplicated coordinates. This has led me to implement a check for duplicated coordinates in an input file. The revised DSSR now reports duplicated coordinates and their corresponding atoms, and it quits if the number of duplicated atoms exceeds a certain threshold. For `R1211TS159_1`, the revised DSSR output would be as below:
1 [e] xyz repeated 1904 times:[0.000 0.000 0.000] 1509-P@0.G1 3412-C6@0.C90
[w] no-of-repeats=1 max-freq=1904
...too many duplicates... quit!
2. `clash`: Both files `R1250TS208_1o` and `R1250TS417_1o` contain multiple models, as visible in PyMOL. Each PDB file uses a single MODEL/END pair to include all its models. This setup is akin to an NMR ensemble but without MODEL/ENDMDL delimiters, which leads to clashes when analyzed together. I have revised DSSR to explicitly check for such clashes and terminate execution if too many are detected. Using `R1250TS208_1o` as an example, the DSSR output would be as below:
[i] 0.G1 and 1.G1 in clashes: min_dist=0.57
[i] 0.G1 and 3.G1 in clashes: min_dist=0.35
[i] 0.G1 and 4.G1 in clashes: min_dist=0.41
...too many clashes... quit!
The above list contains only three of the many clashes detected in this file. One can notice immediately the G1 nucleotides from chains `0`, `1`, `3`, and `4` are in clashes (see the attached file `clashes_208.pdb`, which contains only G1 nucleotides from the four chains).
3. `superimposed`: The five example files (`R1283v3TS304_1o` ... `R1283v3TS304_5o`) have similar issues as the clash cases. Running the revised DSSR on `R1283v3TS304_1o` would produce the following output:
[i] 0.A1 and 2.A1 in clashes: min_dist=0.74
[i] 0.A1 and 3.A1 in clashes: min_dist=0.78
[i] 0.A1 and 4.A1 in clashes: min_dist=0.56
...too many clashes... quit!
Here A1 nucleotides from chains `0`, `2`, `3` , and `4` are in clashes (see the attached `superimpose-1.pdb`).
How the `clash` and `superimposed` categories are supposed to be different? They look similar to me.
Overall, the `error_examples` (in `all_zero_xyz`, `clash`, and `superimposed`) pose problems because they do not contain valid DNA/RNA structures as a whole. DSSR cannot extract meaningful information from these files. However, the revised DSSR explicitly highlights these issues, saving users from spending time on invalid data. Do these DSSR revisions make sense to you?
In the end, I am glad to receive the following feedback from the authors:
Thanks, these revisions all make sense! The examples I sent on clashes and superimposed were actually similar and I think the error output makes sense as well.
Final thoughts
This blog post offers an in-depth look at my efforts to enhance DSSR. As the developer of this software product, I am deeply committed to ensuring its quality and usability. I extend my gratitude to the authors for their valuable feedback and assistance in resolving these issues. In return, the updated version of DSSR (v2.5.4-2025jun04) should not only streamline their workflow but also benefit the broader user community.
For those who read through this lengthy post, I want to emphasize that DSSR is actively supported: I am here to listen and help. Any questions related to its use, bug reports, or feature requests are warmly welcomed on the 3DNA Forum. As I’ve mentioned before, please don’t hesitate to share any negative experiences or bugs with DSSR—just ensure to provide specific details so others can reproduce the issue. I will address these concerns as soon as I’m aware of them and will frankly acknowledge any mistakes I may have made. My goal is for DSSR to be a reliable software tool that the community can trust and build upon.
References
Kretsch,R.C. et al. (2025) Assessment of nucleic acid structure prediction in CASP16. bioRxiv; https://doi.org/10.1101/2025.05.06.652459.
In DSSR, the --frame option allows users to reorient a nucleic acid structure using the standard base reference frame (see Olson et al., 2001). This option can be applied not only to an individual base frame but also a base-pair frame, or the middle frame between two bases or base pairs. These variations facilitate the alignment of nucleic acid structures for a wide range of comparative analyses. In this blog post, I will demonstrate how to use the --frame option with concrete examples, enabling readers to apply this unique DSSR feature to their own projects.
The standard base reference frame
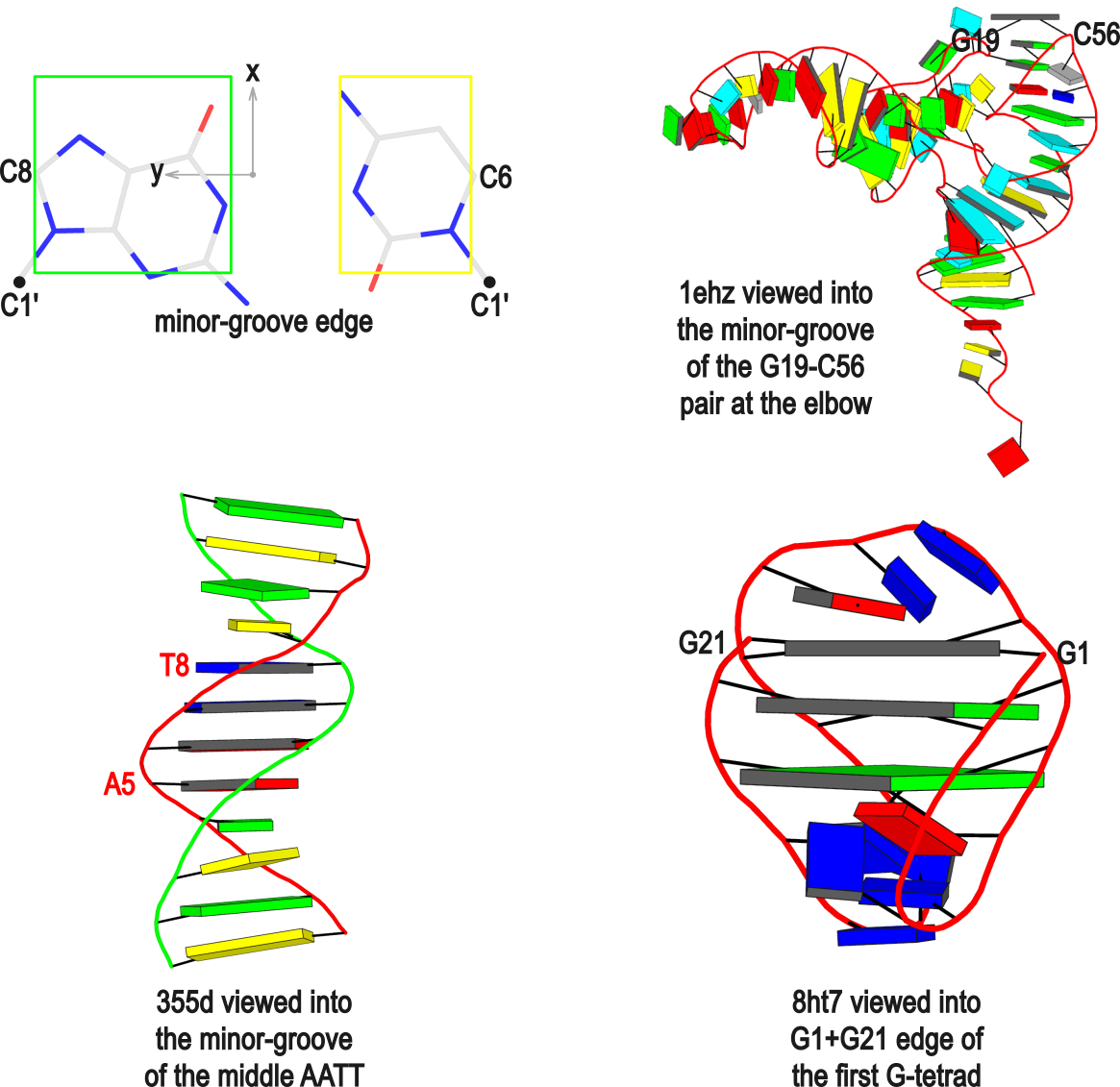
The standard base reference frame is derived from an idealized Watson-Crick base pairing geometry (top-left, figure below). The x-axis points in the direction of the major groove along what would be its pseudo-dyad axis—that is, the perpendicular bisector of the C1'...C1' vector spanning the base pair. The y-axis runs along the long axis of the idealized base-pair in the direction of the sequence strand, parallel to the C1'...C1' vector, and is displaced so as to pass through the intersection between the (pseudo-dyad) x-axis and the vector connecting the pyrimidine Y(C6) and purine R(C8) atoms. The z-axis is defined by the right-handed rule. For right-handed A- and B-DNA, the z-axis accordingly points along the 5' to 3' direction of the sequence strand.
Typical usages of the --frame option
Using the classic B-DNA dodecamer PDB entry 355d as an example, DSSR can be run with the --frame option as follows:
# 1...5..8....
# chain A: 5'-CGCGAATTCGCG -3'
# chain B: 3'-GCGCTTAAGCGC -5'
# reorient 355d in the reference frame of C1 on chain A
x3dna-dssr -i=355d.pdb --frame=A.1 -o=355d-b1.pdb
# reorient 355d in the frame of the Watson-Crick pair C1-G24
x3dna-dssr -i=355d.pdb --frame=A.1:wc -o=355d-bp1.pdb
# ... with the minor-groove of pair C1-G24 facing the viewer
x3dna-dssr -i=355d.pdb --frame=A.1:wc-minor -o=355d-bp1-minor.pdb
# with the minor-groove of the middle AATT tract facing the viewer
x3dna-dssr -i=355d.pdb --frame='A.5:wc-minor A.8:wc' -o=355d-AATT-minor.pdb
# Rendered in cartoon-blocks with base-pair blocks, and black minor-groove
# Load 355d-AATT-minor.pml into PyMOL (bottom-left, figure above)
x3dna-dssr -i=355d-AATT-minor.pdb --cartoon-block --block-file=wc-minor -o=355d-AATT-minor.pml
The abbreviated notation A.1 refers to nucleotide numbered 1 (as indicated in the coordinates file) on chain A. Here, it denotes C1, as shown at the top of the listing. Similarly, A.5 and A.8 correspond to nucleotides A5 and T8 on chain A, respectively. In most cases, such as with 355d, the combination of chain identifier and residue number is sufficient to uniquely identify a nucleotide. More generally, other information such as model number or insertion code may be needed to specify a particular nucleotide.
In the above listing, wc after the colon (for example, A.1:wc) specifies the Watson-Crick base pair that the corresponding nucleotide participates in. Meanwhile, minor transforms the structure so that the minor-groove of the base (or base pair, or step) faces the viewer. The keywords wc and minor are settings that influence the construction or view of the frame. Case or order does not matter for these keywords as long as there is a match—for example, minor+wc works the same as wc-minor.
Two other examples combining the --frame option with cartoon-block representations
The intuitive geometric meaning of the standard base reference frame combined with the DSSR-enabled cartoon-block representation allows for an enhanced understanding of intricate structural features. In the top-right panel of the figure above, we see the classic yeast phenylalanine tRNA (PDB entry 1ehz) viewed into the minor-groove of the pseudo-knotted G19-C56 pair at the elbow of the L-shaped tertiary structure. The stacking interactions of the purines at the top-right of the panel are clearly visible in this view. In the bottom-right panel, an anti-parallel G-quadruplex from PDB entry 8ht7 is shown. The G-tetrads are automatically identified and rendered as square blocks, all with DSSR. This representation makes the chair conformation of the three-layered anti-parallel G-quadruplex crystal clear. The DSSR commands used are listed below:
# yeast tRNA (1ehz)
x3dna-dssr -i=1ehz.pdb --frame=A.19:wc-minor -o=1ehz-elbow.pdb
x3dna-dssr -i=1ehz-elbow.pdb --cartoon-block --block-file=wc-minor -o=1ehz-elbow.pml
# anti-parallel chair-shaped G-quadruplex (8ht7)
x3dna-dssr -i=8ht7.pdb --select=nts -o=8ht7-nts.pdb # extract nucleotides, ignore amino acids
# reorient 8ht7 in the frame of the G-tetrad involving G1, in edge view
x3dna-dssr -i=8ht7-nts.pdb --frame=A.1:G4-minor -o=8ht7-Gtetrad.pdb
x3dna-dssr -i=8ht7-Gtetrad.pdb --block-cartoon --block-file=G4-minor -o=8ht7-Gtetrad.pml
References
Olson,W.K. et al. (2001) A standard reference frame for the description of nucleic acid base-pair geometry. Journal of Molecular Biology, 313, 229–237.
The 3DNA suite includes the mutate_bases program, which, as its name suggests, mutates bases while maintaining the backbone conformation. This feature was incorporated into the suite following user feedback and has been utilized in several studies before being formally published in the Li et al. (2019) paper. A key advantage is that the mutation process preserves both the geometry of the sugar-phosphate backbone and the base reference frame, encompassing position and orientation. Consequently, re-analyzing the mutated model yields identical base-pair and step
parameters as those of the original structure.
In DSSR, the standalone mutate_bases program has become the mutate sub-command with enhanced functionality and improved usability, as documented in the User Manual. The mutate module allows users to perform base mutations efficiently and effectively by taking advantage of the powerful DSSR analysis engine.
To further expand the modeling capabilities of the DSSR, v2.5.3 introduced the --mutate-type option to allow for backbone mutations, based on the base reference frame. Furthermore, the target can be any fragment, regardless of length or composition, rather than just a single nucleotide. When combined with the rebuild module, this feature significantly enhances DSSR’s ability to model nucleic acid structures.
Here is an example of modeling PDB entry 1msy, a 27-nt structure (1msy.pdb) that mimics the sarcin/ricin loop from E. coli 23S ribosomal RNA.
x3dna-dssr analyze -i=1msy.pdb --ss --rebuild -o=1msy-expt.out
mv dssr-ssStepPars.txt 1msy-step.txt
x3dna-dssr rebuild --backbone=RNA --par-file=1msy-step.txt -o=1msy-step.pdb
x3dna-dssr -i=1msy.pdb --select-resi='A 2654' -o=1msy-A2654.pdb
x3dna-dssr -i=1msy.pdb --select-resi='A 2655' -o=1msy-G2655.pdb
x3dna-dssr -i=1msy-A2654.pdb --frame=2654 -o=frame___A.pdb
x3dna-dssr -i=1msy-G2655.pdb --frame=2655 -o=frame___G.pdb
x3dna-dssr mutate -i=1msy-step.pdb --entry='num=8 to=A; num=9 to=G' -o=1msy-C2endo.pdb --mutate-part=whole
x3dna-dssr --connect-file -i=1msy-C2endo.pdb -o=1msy-C2endo-cnt.pdb --po-bond=5.0
- The
analyze step uses options --ss and --rebuild to generate the file dssr-ssStepPars.txt (containing base-step parameters), which is then renamed to 1msy-step.txt. The rebuild step employs 1msy-step.txt to construct a structure (1msy-step.pdb) with regular C3'-endo sugar RNA backbone conformation. Note that the rebuilt structure has nucleotides numbered from 1 to 27, while in the PDB 1msy, they correspond to 2647 to 2673, respectively.
- However, the A2654 and G2655 dinucleotides in 1msy are actually in C2'-endo sugar conformation, creating the S-shaped structure around the GpU platform. The above rebuilt structure does not reflect this distortion. So we extract A2654 and G2655 with
--select-resi and then put each in its standard base reference frame, named frame___A.pdb and frame___G.pdb, respectively.
- Now we mutate A8 and G9 in the rebuilt structure
1msy-step.pdb to A and G with option --mutate-part=backbone to ensure the backbone conformations are changed according to those in frame___A.pdb and frame___G.pdb, respectively. The resulting structure is named 1msy-C2endo.pdb. Now the S-shape around the GpU platform is preserved, even though the backbone are not always covalently connected, due to large O3'(i-1) to P(i) distances between neighboring nucleotides. The last step is to generate CONECT records with --connect-file option to connect the backbone atoms explicitly, resulting in more smooth backbone cartoon representation in PyMOL as shown below.
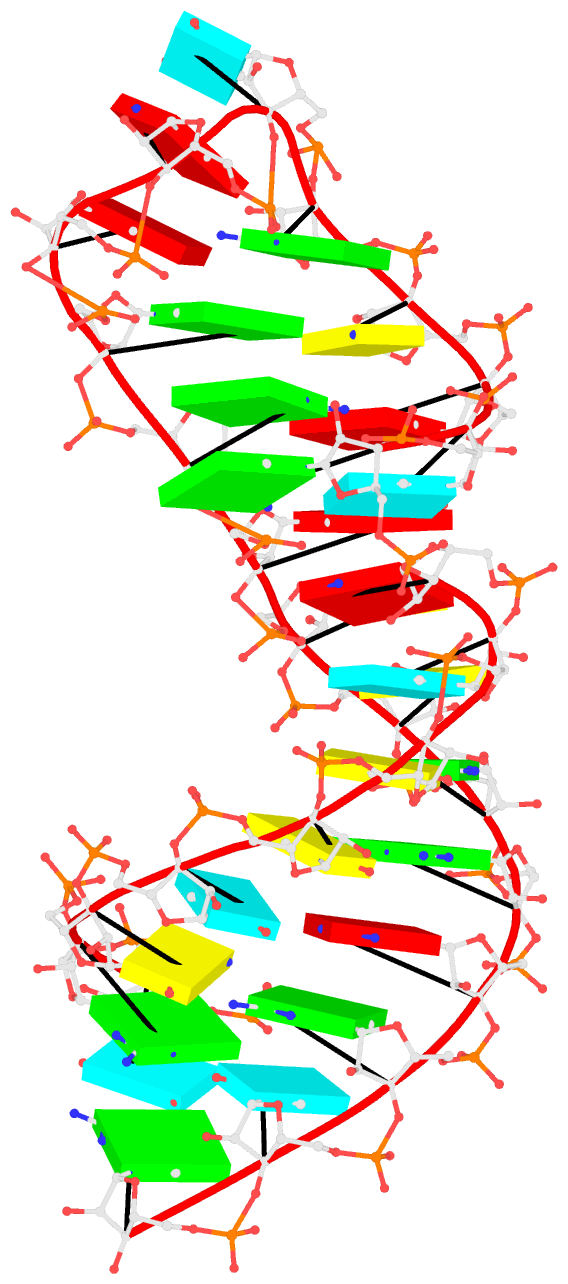
As noted in the Li et al. (2019) paper, users can optimize this approximate backbone connection using Phenix, while keeping the base atoms fixed. The 3DNA-Phenix combination leads to a model where the base geometry strictly follows the parameters prescribed in the user-specified file, and the backbone is regularized with improved stereochemistry and a ‘smooth’ appearance in ribbon representation.
There are other variants of the DSSR mutate module, including for building Z-DNA backbones. However, the above example is sufficient to demonstrate the power of the integrated approach enabled by DSSR for the analysis and modeling of nucleic acid structures. See the DSSR User Manual for more details.
References
Li,S. et al. (2019) Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Nucleic Acids Res., 47, W26–W34.
Recently, I read carefully the two papers by Farag et al. on the ASC-G4 algorithm to calculate "advanced structural characteristics of G-quadruplexes" (2023), and the comprehensive analysis results of intramolecular G4 structures in the PDB (2024). By developing a convention to orient and number the four strands, ASC-G4 allows for unambiguous determination of the intramolecular G4 topology. It also has an in-depth discussion on assigning syn or anti glycosidic configuration of guanosines, and categorizes four different types of snapbacks.
I am glad to see that DSSR is cited in these two papers, as quoted below:
X3dna-DSSR (19) (http://x3dna.org) is a website that was created to calculate nucleic acid structural parameters, like the local base-pair parameters, local step base-pair parameters, torsion angles, etc, but not the special characteristics of G4. A subdomain dedicated to G4, DSSR-G4DB (Dissecting the Spatial Structure of RNA – G4 Data Base) (http://g4.x3dna.org) emanated from this website. It is a database that gathers and calculates some specific structural information about published G4s, like the topology, the rise, the helical twist, etc, but not the groove widths or the presence of snapbacks. -- Farag et al. (2023)
Indeed, DSSR-G4DB dose not classify snapbacks. I was aware of such non-canonical G4s when I first developed the G4 module in DSSR around 2017-2018, and the V-shaped loops was derived to reflect the peculiarity of snapbacks.
DSSR classifies groove widths as medium, wide, or narrow, based on the glycosidic angles of neighboring guanosines in a G-tetrad, following the G4 literature. Using PDB entry 2lod as an example, the relevant part of the DSSR output is shown below. The groove widths of the three G-tetrads in the G4-stem have the same pattern of groove=--wn, standing for medium, medium, wide, and narrow, respectively. Note that the medium groove is represented by a dash instead of m because --wn stands out more clearly than mmwn (similar idea applies to glycosidic bond, e.g., sss-).
1 glyco-bond=sss- sugar=---- groove=--wn Major-->WC N- nts=4 GGGG A.DG1,A.DG6,A.DG20,A.DG16
2 glyco-bond=---s sugar=---- groove=--wn WC-->Major N+ nts=4 GGGG A.DG2,A.DG7,A.DG21,A.DG15
3 glyco-bond=---s sugar=---- groove=--wn WC-->Major N+ nts=4 GGGG A.DG3,A.DG8,A.DG22,A.DG14
Since DSSR-G4DB is a database, the user cannot provide his own G4 structure, to obtain structural information. Hence the necessity of developing a website where the user uploads his G4 structure file to obtain all its important and specific structural characteristics (like the topology, the groove width, the tilt and twist angles, etc.). This can be very useful, not only for the analysis of published PDB structures but also for structures in refinement or obtained from MD simulations, to evaluate their quality. To our knowledge, there is no website dedicated to G4 to do such calculations in real-time. Therefore, we developed the algorithm ASC-G4 (advanced structural characteristics of G4) and deployed it as a user-friendly website at the following address: http://tiny.cc/ASC-G4. -- Farag et al. (2023)
Thanks to the NIH R24GM153869 grant support, the http://g4.x3dna.org website now allows users to upload their own atomic structures in PDB for mmCIF format for the identification, annotation, and visualization of G4s. See the example of uploading PDB coordinate file 2lod.pdb.
As background, I had long aspired to develop a dynamic website for on-demand G4 structural analysis but was unable to pursue this goal until recently. During the 4-year funding gap, I still managed to maintain the website g4.x3dna.org, which provides DSSR results for G4 structures in the PDB (a resource now known as the DSSR-G4DB database). To date, the only published work related to G4s is my 2020 paper on the integration of DSSR with PyMOL. Clearly, a dedicated method paper detailing the G4 module in DSSR and the g4.x3dna.org website has been long overdue.
As an initial step toward addressing this gap, I have recently revised the G4-related code in DSSR, fixed existing bugs, and added new features. The g4.x3dna.org website has undergone a complete overhaul, enabling users to upload their own structures for dynamic G4 analysis. Additionally, the DSSR-G4DB database is being actively updated on a weekly basis as new PDB entries are added.
Calculation of the twist and tilt angles. In G4, the helix twist is the rotation of a tetrad relative to its successive one. To measure the twist angle, the most spread method is that described by Lu and Olson (2003) (32) and Reshetnikov et al. (2010) (33). In this method, the angle is calculated from the dot product between two C1’–C1’ vectors from two successive tetrads, i and i + 1, the C1’ atoms of each vector belonging to two adjacent guanosines of a Hbp. The issue with this method is that it does not allow access to the sign of the angle, which defines the direction of the G4 helix, viz. right-handed or left-handed. -- Farag et al. (2023)
There is clearly a misunderstanding in the above text. 3DNA/DSSR can handle left-handed Z-DNA without any issues. DSSR also reports negative twist angles for left-handed G4s, as shown clearly for PDB entry 7d5e, for example.
3DNA/DSSR derives a complete of set of six base-pair parameters (including shear and opening), six step parameters (including twist and rise), and six helical parameters, using a rigorously defined and completely reversible algorithm (CEHS) and the standard base-reference frame. See section "3.2.3 Base pairs" in DSSR User Manual for more details. The DSSR output for G4s (as in DSSR-G4DB) reports only twist and rise, along with overlapped areas, simply because these are the most important parameters and easily interpretable.
The list of the resolved G4 structures was downloaded from the ONQUADRO website (https://onquadro.cs.put.poznan.pl/) (39) at about the end of October 2023. It consisted of 291 intramolecular structures (named unimolecular in the website) and 154 intermolecular G4s (96 bimolecular and 58 tetramolecular). Only the intramolecular structures were kept for this study. To this list, we added 55 missing intramolecular structures that were found on the website of DSSR-G4DB (http://g4.x3dna.org) (40). From the merged list, 345 structures were downloaded from the Protein Data Bank (PDB) (http://www.rscb.org/pdb/) (41) because one structure had no available coordinates in the PDB format (7ZJ5 (42)). -- Farag and Mouawad (2024)
DSSR adopts the frame of reference of Webba da Silva, designating the four strands and grooves of G4-stem as shown below using PDB entries 8ht7 (G1 in syc) and 5ua3 (G1 in anti) as example for the syn or anti glycosidic bond of the 5'-guanosine, respectively.
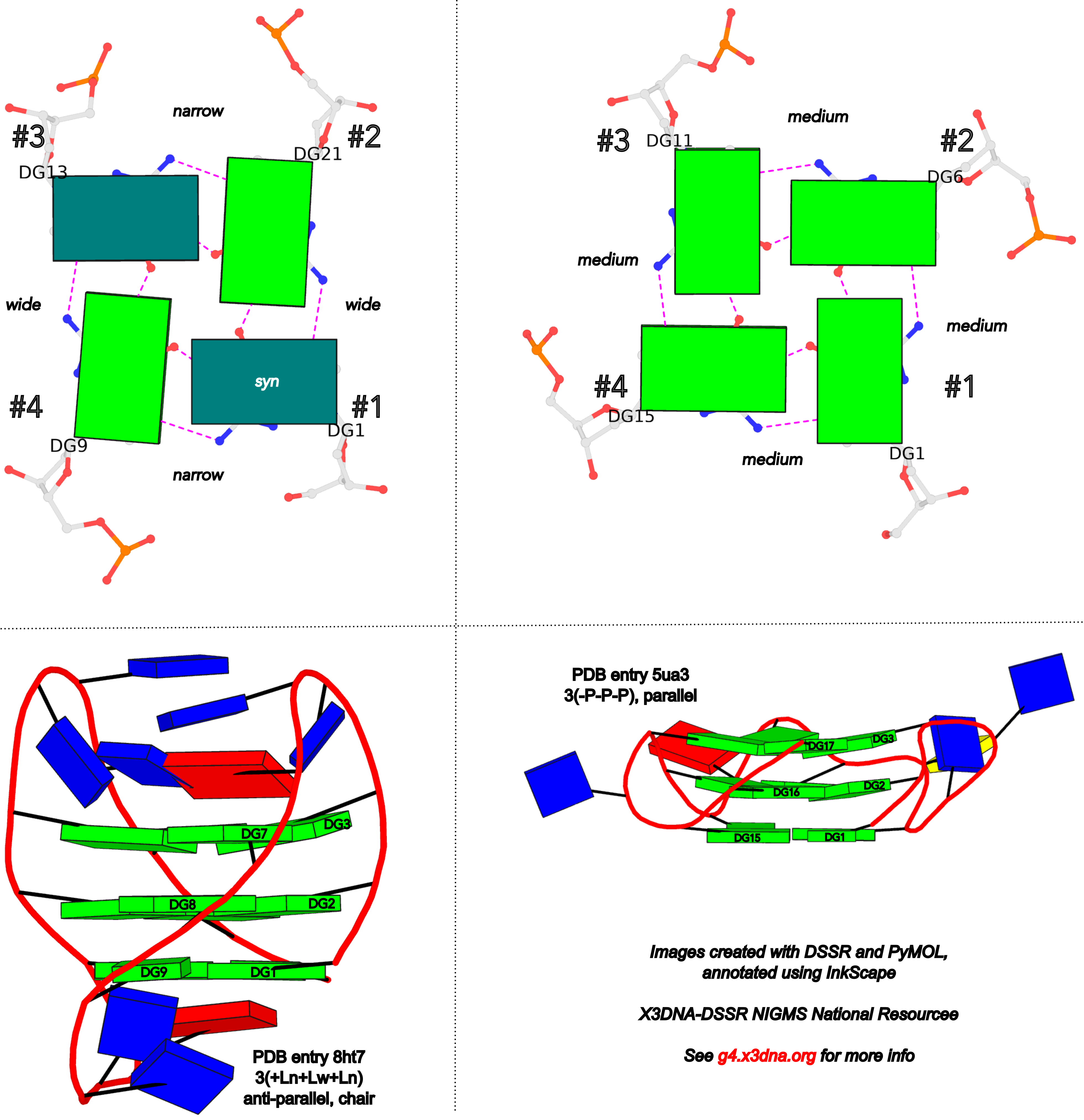
In DSSR, the first strand (#1) is always upward (U) from 5' to 3'-end, and the polarity of the other three strands is determined by its orientation relative to #1: U if parallel, or D if antiparallel. There are a total of 2x2x2=8 possible combinations of U and D for the three strands, which define parallel (U4: UUUU), antiparallel (U2D2: UDDU, UDUD, UUDD), or hybrid (UD3: UDDD; U3D: UDUU, UUDU, UUUD). For example, the PDB entry 2lod is characterized by DSSR as: "hybrid-(mixed), UUUD, U3D(3+1)", and PDB entry 8ht7 as: "anti-parallel, UDUD, chair(2+2)". This notation is topologically equivalent to the one adopted by ASC-G4 but with opposite orientation of the strands.
Overall, DSSR and ASC-G4 provide different perspectives on G4 structures. It is to the user to decide which one is more suitable for their needs.
References
Farag,M. et al. (2023) ASC-G4, an algorithm to calculate advanced structural characteristics of G-quadruplexes. Nucleic Acids Res., 51, 2087–2107.
Farag,M. and Mouawad,L. (2024) Comprehensive analysis of intramolecular G-quadruplex structures: furthering the understanding of their formalism. Nucleic Acids Res., gkae182.
In late September of 2018, I contacted Dr. Mateus Webba da Silva requesting a copy of his 2007 article, titled "Geometric formalism for DNA quadruplex folding". At that time, I had implemented a G4 module within DSSR for the automatic identification, comprehensive annotation, and schematic visualization of G-quadruplexes from 3D atomic coordinates. I noticed the 2007 paper, and was intrigued by the following sentences in the abstract:
A formalism is presented describing the interdependency of a set of structural descriptors as a geometric basis for folding of unimolecular quadruplex topologies. It represents a standard for interpretation of structural characteristics of quadruplexes, and is comprehensive in explicitly harmonizing the results of published literature with a unified language.
Mateus kindly sent me a copy of the 2007 article, and shortly afterwards he also shared with me the Dvorkin et al. (2018) paper on "Encoding canonical DNA quadruplex structure". I carefully read both papers, plus the Karsisiotis et al. (2013) tutorial paper. I was impressed by the elegance of the formalism: simple and systematic, so I immediately decided to add this feature to the G4 module of DSSR.
As the Chinese saying goes, "纸上得来终觉浅,绝知此事要躬行" ("What you learn from books is always shallow. You must practice it yourself to know it well." -- Google Translate). The implementation process was challenging because of subtleties in the formalism, but very rewarding. It is all about scientific understanding and software engineering. Only after a thorough understanding and attention to meticulous details can one create a robust and reliable software tool. On the other hand, once properly implemented, the DSSR G4 module can be applied consistently. Any discrepancies between DSSR output and literature merit further investigation. These discrepancies could either arise from bugs in DSSR (which I will promptly address upon identification) or, more likely, typos or errors in the reported results.
Webba da Silva (2007) systematically described the interdependency of glycosidic bond (syn or anti), strand polarity (parallel or anti-parallel), groove width (narrow, medium, or wide), and loop type (lateral, propeller, or diagonal) in unimolecular G-quadruplexes. Figures 1-3 and Scheme 1 of Webba da Silva (2007) are very informative, and easy to follow conceptually. The Karsisiotis et al. (2013) tutorial provided further details based on experimentally determined G-quadruplex structures from the PDB (e.g., Figure 3: the schematic for all possible combinations of glycosidic bond and the corresponding groove-width combinations in G-tetrad). Some key observations:
- Since glycosidic bond can be either syn or anti, there a total of
2x2x2x2 = 16 possible combinations in a G-tetrad.
- The disposition of glycosidic bond of guanosines in a G-tetrad leads to only eight possible groove-width combinations.
- Only tetrads with the same groove-width combinations may stack to form stable G-quadruplexes.
- Propeller loops invariably link medium grooves within a G-quadruplex stem.
- Lateral and diagonal loops bridge guanosines of different glycosidic bond.
- If a single-stranded quadruplex starts with a narrow groove, it can only be with a clockwise loop progression (i.e., +lateral).
- There are 26 permissible looping combinations within a canonical unimolecular G-quadruplex (G4-stem).
To unambiguously characterize a G4-stem, Webba da Silva (2007) defined a frame of reference where the 5’-G in a G4-stem is set as the origin, and the first strand is progressing towards the viewer. Regardless of the clockwise or anti-clockwise progression of the base sequence, the scheme designates one orientation for the syn and anti glycosidic bond by following G+G H-bonding alignments. Put another way, grooves and strands are strictly related to the reference (first) strand in an anti-clockwise manner, irrespective of the progression of the base sequence. The point is illustrated in the figure below, using PDB entries 8ht7 (G1 in syc) and 5ua3 (G1 in anti) as an example for the syn or anti glycosidic bond of the 5'-guanosine, respectively.
Based on previous work, the Dvorkin et al. (2018) paper proposed a systematic nomenclature for G4-stem. The single structural descriptor contains:
- The number of G-tetrads (i.e., the G-tract length).
- Loop types (lowercase l for lateral, p for propeller, and d for diagonal) and relative direction ("+" for clockwise and "-" for anti-clockwise progression, using the frame of reference described above).
- For lateral loops, the groove widths ("w" for wide, and “n” for narrow) are denoted in subscript.
So a complete descriptor could be 2(+lnd−p), as shown in Figure 1A of the Dvorkin et al. (2018) paper. Significantly, Figure 1B therein further gave structural descriptors for six experimentally determined G4-stems from the PDB. These examples, plus the ones in the supplementary materials, were used to validate my implementation of the systematic nomenclature in the G4 module of DSSR. My results agree with those in the Dvorkin et al. (2018) paper, except for two cases, which are discussed below.
- For PDB entry 2gku: 3(-p-ln-lw) (Dvorkin et al.) vs 3(-P-Lw-Ln) (DSSR), with swapped n (narrow) and w (wide) groove widths for both lateral loops.
- For PDB entry 2lod: 3(-pd+ln) (Dvorkin et al.) vs 3(-PD+Lw) (DSSR), with swapped n (narrow) and w (wide) groove width for the lateral loop.
Note that in DSSR, I am using uppercase L/P/D for lateral/propeller/diagonal loop types, and lowercase n/w for narrow/wide groove widths, respectively. Doing so distinguishes between the different loop types and groove widths in pure text format.
After careful examination of these discrepancies, I still couldn’t find any errors in my implementation. So I contacted Mateus for verification (in early October 2018). Thankfully, he quickly responded and acknowledged the mistakes for PDB entry 2gku in Dvorkin et al. (2018), saying "There can not be a –Ln after the –p." Clearly, the wrong descriptor for PDB entry 2gku in Dvorkin et al. (2018) was due to a typographical error. This example illustrates the power of a robust software tool like DSSR.
References
Dvorkin,S.A. et al. (2018) Encoding canonical DNA quadruplex structure. Sci. Adv., 4, eaat3007.
Karsisiotis,A.I. et al. (2013) DNA quadruplex folding formalism – A tutorial on quadruplex topologies. Methods, 64, 28–35.
Webba da Silva,M. (2007) Geometric formalism for DNA quadruplex folding. Chemistry A European J, 13, 9738–9745.